Frequently asked questions
Products & Services
Antibodies
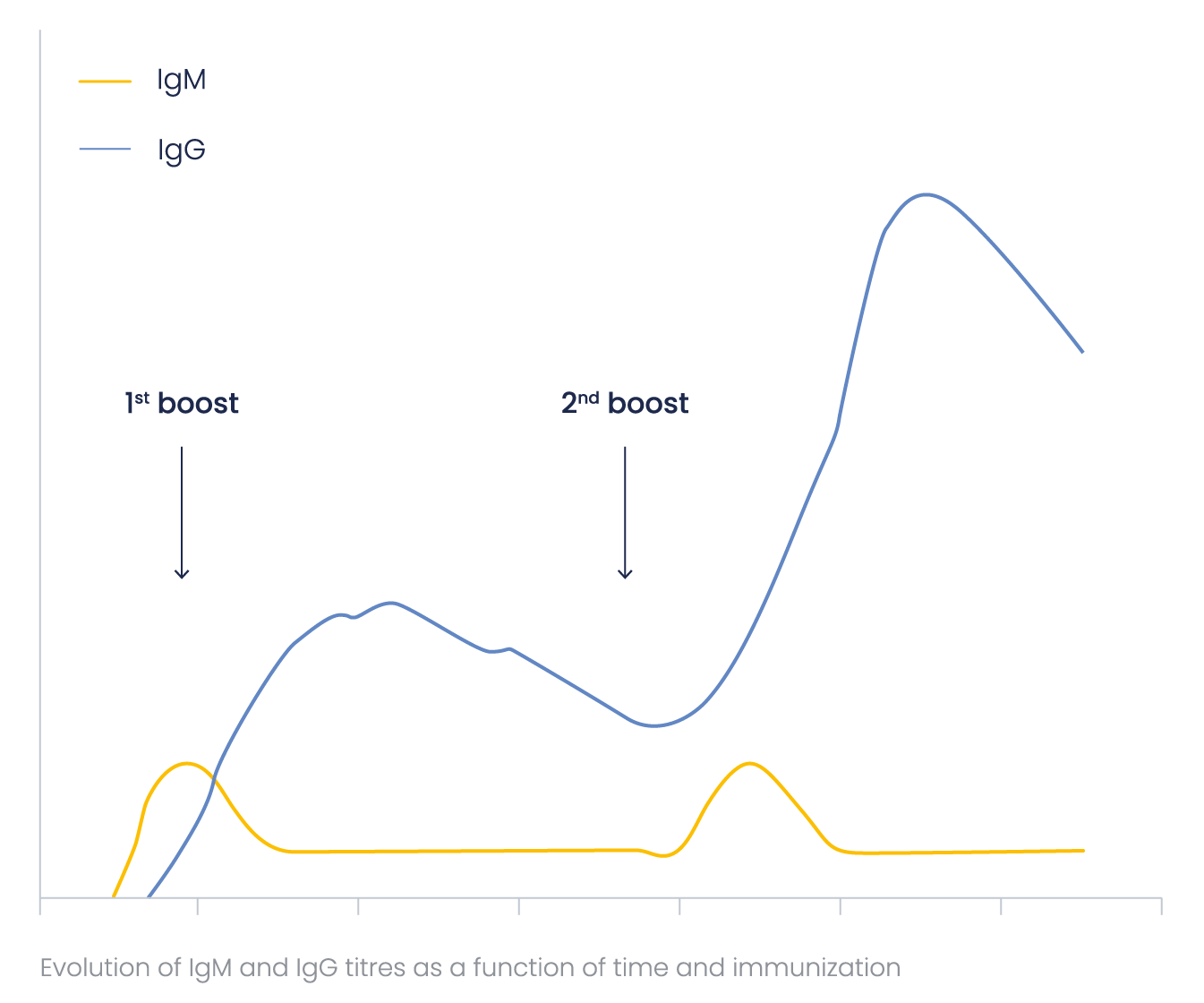
How does antibody titer evoluate?
IgMs are more difficult to label and detect than IgGs because of their pentameric structure. To obtain the optimal (highest) IgG/IgM ratio, Eurogentec’s immunization programs are optimized concerning the number and frequency of the boosts.
Special attention was paid to this ratio when developing the Speedy 28-day program, to ensure a minimal amount of IgMs in the final bleed. IgM and IgG antibody titer as a function of time may be represented in the general scheme below.
What are the expected bleed volumes?
The immune response depends on the antigen but also on the host. Large animals provide the advantage of allowing larger bleeds but their immune response is generally lower than small animals. The following table indicates the expected bleed volume in various hosts.
| Pre-immune | Small Bleed | Large bleed |
Final bleed | Comment | |
| Mouse | 40-70 μL | 40-70 μL | 40-70 μL | 300-500 μL | Good to test antigenicity |
| Guinea pig |
1 mL | 1 mL | 2-3 mL | 10-15 mL | For small serum volumes |
| Rat | 2 mL | 2 mL | 2 mL | 5 mL | For small serum volumes |
| Chicken | 1 egg | ± 8-10 eggs* | ± 8-10 eggs* | ± 8-10 eggs* | For mammalian antigens and large quantities of Antibodies, 4 eggs = |
| Rabbit | 2 mL | 2 mL | 20-25 mL | 50-70 mL | For most applications |
| Goat | 2 mL | 2 mL | 250 mL | 1000 mL | For large batch volumes |
| Sheep | 2 mL | 2 mL | 250 mL | 1000 mL | For large batch volumes |
| Llama | 2 mL | 2 mL | 250 mL | 1000 mL | Single chain antibodies and for large batch volumes |
How much Antigen do I need for an immunization?
Injection amounts per rabbits depend on the antigen weight:
100 μg per injection for < 18 – 20 kDa proteins*;
200 μg per injection for > 18 – 20 kDa proteins.
Check here the Injection amounts for other hosts.
*Please note that the molecular mass of haptens is usually too low to elicit an immune response. Therefore, they should be provided under a format with a higher MM if Ab production is requested (e.g. coupled to a carrier).
Which animal should I use for antibody production?
The choice of animal is dependent on practical factors such as the source of the antigen, quantity of serum required for research, the titer of the antibody and the time required to generate antibodies. Antigens coming from rabbits should be injected into pigs or chickens rather than into rabbits. Mammalian proteins can be injected into chickens to provide high titer antibodies, which have the advantage of being IgY type antibodies.
Program duration: generally, the smaller the animal the shorter the program duration and the smaller the volumes of serum received. As antigenic response varies from animal to animal of the same species, repeating a immunization program is more risky than choosing a larger animal for antibody production to ensure sufficient material. It is recommended to immunize at least 2 animals to ensure a high quality antibody at the end of the program.
Which animals are available?
Eurogentec offers a very large range of animals, mice, rats, chickens, rabbits, sheep, goats, pigs, llamas, alpacas... Should you require a different host than listed please let us know.
What is an SPF animal?
Specific Pathogen Free animals are bred uniquely for the production of antibodies. These animals are housed in a pathogen free environment – all entering air, food, water is guaranteed to not contain foreign organisms that could provoke an immune response. These animals offer a very low background signal due to the restricted environment in which they live.
How long does antibody production take?
Antibody production can be done in as short a time as 2 months although we recommend 3 months for most hosts.
How many boosts and bleeds do you do?
The immunization program is species dependent, however, in general animals are immunized three times the first month then are given booster shots once a month for as long as is required. Bleeds are done 15 days after immunizations.
What happens if you reuse an animal for a second or third immunization?
The risk of reusing animals for other immunization programs is the presence of antibodies from past immunizations. This can lead to increased background signals and false positive responses in downstream applications.
Is it important to get a terminal / final bleed?
Yes, terminal bleeds ensures that you are not getting serum from a host that has already been used in a previous immunization program, which could potential contaminate your supply of antibodies.
What if my antigen is insoluble in biological buffers?
Protein concentrations should be at least 1mg/ml with minimal amounts of urea, SDS, guanidine hydrochloride (and other chemotropic agents). Should the antigen require the presence of these agents then the antigen concentration should be raised to 5-10 mg/ml. By injecting small volumes (10-50 µl) we are often able to generate an immune response to the antigen.
What buffers are compatible with immunizations?
Most biologically related buffers are compatible with immunization, this includes PBS, Tris buffer, phosphate buffer, in moderate molarity. The antigen should be in a concentration of 1mg/ml or better and should be shipped frozen on dry ice when possible.
How should I ship my antigen?
Samples should be shipped on dry ice whenever possible. Peptides and SDS-PAGE gel slices can be sent at room temperature.
Can I extend my program to get more bleeds and antibodies?
All programs can be extended by adding additional immunizations and monitoring response by ELISA. When sufficient titer is present the final bleed can be ordered.
What is the pre-immune serum for?
Evaluation of pre-immune serum in your application permits the selection of the best animal for the subsequent production of antibodies. The selection criterion is no-signal in the application (eg nothing in Western Blot) due to the presence of previously generated antibodies thus ensuring very low background signal after immunization. Pre-screening is particularly important when making antibodies to against common bacterial, viral or common allergens. We recommend screening 5 or 10 animals and selecting the best for immunization.
How long should my peptide be?
Epitopes are generally 6-8 amino acids in length, by using a peptide of 16 amino acids in length there is the possibility of raising 10 different antibodies against the sequence used. This elevates the success rate of the program, although we have had success using peptides of shorter and longer lengths. Long peptides (>20’mers) are not recommended as these could contain secondary structures not found in the natural antigen.
Epitopes are generally 6-8 amino acids in length, by using a peptide of 16 amino acids in length there is the possibility of raising 10 different antibodies against the sequence used. This elevates the success rate of the program, although we have had success using peptides of shorter and longer lengths. Long peptides (>20’mers) are not recommended as these could contain secondary structures not found in the natural antigen.
How is the peptide conjugated to the carrier protein?
Peptides can be conjugated to a carrier protein via the N-terminus, C-terminus or an internal amino acid (not recommended). Coupling with glutaraldehyde is used to couple the N-terminus of the peptide to the lysine residues (via the amino sidechain) of the carrier protein; coupling with EDCI (1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide) is used to link the C-terminus of the peptide to the lysine residues of the carrier protein; coupling with MBS (m-Maleimidobenzoyl-N-hydroxysuccinimide ester) is used to link a cysteine via its SH group to the lysine residues of the carrier protein. This very common approach involves adding a cysteine residue to the selected peptide sequence prior to coupling.
What is a carrier protein?
A carrier protein is a protein that is used to deliver the peptide in an immunization program. As the peptide’s molecular weight of 1.5 – 2 kDa will not elicit an immune response on its own, the peptide is conjugated to a protein to increase its apparent molecular weight. The host will then generate antibodies against the peptide and the carrier protein.
Which carrier protein should I use?
For best results a carrier protein should elicit a strong immune response. For this reason the carrier protein should be unrelated to the host species.
Eurogentec offers a number of carrier proteins (BSA, OVA, THY), but we find that we get best results from KLH (Keyhole limpet hemocyanin).
What purity of peptide is best for antibody production?
Although high purity peptides are better than impure peptides for the generation of specific antigens, high purity peptides are not required for successful antibody generation. Immunograde peptides have greater than 70 % purity and are routinely used in immunization programs.
Is it possible to make antibodies against non-peptides (haptens)?
In analogy to conjugating peptides, it is possible to conjugate low molecular weight molecules to spacers and then to carrier proteins. In this way antibodies against hormones for example can be generated.
What are MAP peptides?
Multiple Antigen Peptide is an alternative method of generating antibodies from peptides. Rather than conjugating the desired peptide to a carrier protein the peptides are synthesized onto a polylysine resin. The resulting structure, once cleaved from the resin, is a polylysine core surrounded by 8 copies of the peptide with an overall molecular weight of 13-17 kDa, sufficient to elicit an immune response. Literature reports suggest that better immune responses are generated from this approach in some cases.
Purification of antibodies generated by the MAP strategy require the additional synthesis of free peptide for subsequent affinity purification.
What kinds of antibody purifications are available?
There are two main types of purifications; the first is to concentrate the antibodies from the other proteins found in serum, the other is to specifically concentrate the antibody of interest. The first case can be done with protein A or protein G columns. Protein A columns are useful for concentrating polyclonal antibodies from human, rabbit, horse, pig, guinea pig, dog while protein G columns are useful for concentrating polyclonal antibodies from mouse, rat and goat sources. Affinity purification to solely concentrate the antibody of interest can be done by coupling the peptide or the purified protein to a resin. The affinity of the antibody for the antigen will cause the retention of the desire antibody. This is then eluted with a salt or pH gradient.
Is there a way to monitor antibody production during the immunization program?
A common approach for evaluating the presence of the desired antibody is with an ELISA (Enzyme Linked ImmunoSorbent Assay). This involves the coating of a plate with the desired peptide, capturing the relevant antibodies from the serum, washing to retain only the relevant antibodies, detecting with the secondary antibody HRP (horse radish peroxidase) in the presence of a chromophore. This colorimetric assay is used to determine the titer of antibody present in the serum. Eurogentec recommends doing an ELISA after the first bleed.
What happens if the ELISA results are negative?
Negative results are replicated to ensure that the results are reproducible. Confirmation of results is best done by Western blot analysis to confirm the absence of antibodies. In the case of both negative ELISA and Western blot results we would recommend the termination of the program and the selection of a different antigen and/or a different host.
What is an ELISA test?
A common approach for evaluating the presence of the desired antibody is with an ELISA (Enzyme Linked ImmunoSorbent Assay). This involves the coating of a plate with the desired peptide, capturing the relevant antibodies from the serum, washing to retain only the relevant antibodies, detecting with the secondary antibody HRP (horse radish peroxidase) in the presence of a chromophore. This colorimetric assay is used to determine the titer of antibody present in the serum. Eurogentec recommends doing an ELISA after the first bleed.
What is a secondary antibody?
A secondary antibody is an antibody used to detect the presence of the primary antibody. An antigen is used to generate a primary antibody in a host animal, for example in rabbit. To detected and quantify the amount of the primary antibody (ELISA, WB, etc) a secondary antibody is used, in this example an anti-rabbit antibody. Typically, the secondary antibody is conjugated to an enzyme that converts a substrate from a non-colored state to a colored state, providing a colorimetric assay. When used with standard curves one can accurately quantify the amount of primary antibody present in a sample.
How many egg yolks does a standard program yield?
Chickens typically yield an egg a day. A 2 month protocol thus yields approximately 30 eggs, the first month being the month of immunizations.
What is Western blotting?
Is a qualitative technique to detect the presence of an antigen in a sample with an antibody. The process involves extracting all the proteins from a sample, separating the proteins in 1D or 2D polyacrylamide gels, transferring the proteins from the gel to a membrane (PVDF or nitrocellulose), blocking the portions of the gel where there are no proteins (typically with milk), treating the membrane with the antibody, washing away excess antibody, detecting the presence of the antibody with a secondary antibody. This method is dependent on the epitope the antibody recognizes. As gel separation methods are denaturing the best antibodies for Western blotting are those that recognize linear sequences.
What causes background in a Western blot?
Background signals can due to non-specific binding of the primary antibody to proteins other than the antigen; due to the presence of spurious antibodies mixed in with the primary antibody; or poor blocking of membranes.
How should I store my antibody?
Storage Suggestion for Antibodies used for in vitro Research Applications
Properly stored, antibodies should remain stable for months or years. Freezers with automatic de-frosting are absolutely inappropriate for antibody storage.
If you are using catalogue antibodies, please refer to the provided data sheet.
Storage Temperatures
Our purified antibodies are delivered to you in PBS with 0,1% BSA as stabiliser and 0,01% Thimerosal as preservative. For these formulations we suppose that storage of small aliquots at –20°C is appropriate.
The aliquoting of antibodies has the following advantages:
- Minimisation of the damage due to freezing and thawing
- Exclusion of contamination due to multiple pipetting from a single vial
Aliquots should be thawed once, with any remaining antibody kept at 4°C.
When you receive an antibody you should carefully thaw it (in the fridge in a 4°C water bath – do not forget about it) and centrifuge it with 10.000 x g for 20 sec in order to pull down all the solution. Aliquot the antibody in low-protein-binding micro centrifuge tubes for storage. Please adapt the size of the aliquots to your typical experimental consumption. Aliquots should not be smaller than 10 µl to avoid damage of the antibody by evaporation and adhesion to the vessel wall.
Though it is considered in general as optional, we recommend the addition of 50% glycerol (v/v, final) to your antibodies for additional freezing and evaporation protection for storage at –20°C (glycerol is frozen at –80°C).
Please use just sterile chemicals and solutions for your antibodies to prevent down stream microbial growth.
Upon receipt, antibody storage for one or two weeks (time required for experimental establishment) at 4°C should be acceptable with subsequent aliquoted freezing for long-term storage.
Ascites and serum represent an exception here since the contained proteases require immediate storage at -20°C or -80°C.
Please be advised that sera produced at Eurogentec do not contain any preservative, 0,01% azide or thimerosal (depending on your research purpose) should be added to the aliquots for storage at –20°C after first thawing.
Egg yolks in contrast must never be frozen, since they denature into a form that does not allow isolation of IgY and affinity purification anymore. Egg Yolks from Eurogentec are shipped at 4°C with 0,01% azide added as preservative ready for short-term storage (over several weeks) at 4°C.
For long-term storage we recommend the purification of IgY from the egg yolks.
IgY isolates and affinity purified IgY antibodies should be treated like purified antibodies described in the beginning of this section.
Antibody formulations that require special attention:
- Enzyme-conjugated antibodies should not be frozen at all in order to retain the maximum enzymatic activity, storage at 4°C is therefore mandatory, please refer to the technical datasheet on the solvent formulation, and add if required 0,01% Thimerosal (Azide will inactivate HRP-conjugates);
- All conjugated antibodies (enzymes, fluorophores, or biotin) should be stored in the dark (very well suited: the black foil bags of x-ray films or photographic paper, and the corresponding boxing);
- Fluorophore-conjugated antibodies can be stored aliquoted at –20°C with 50% glycerol (final v/v) – please make sure by the data sheet that they are formulated as well with BSA, physiologically concentrated buffer salts (e.g. PBS), and preservatives (azide or thimerosal), furthermore, please be sure to use a sterile glycerol formulation, since microbial growth might occur and destroy the antibody. Storage at –80°C of glycerol containing solutions is not advised since this is below the freezing point of glycerol;
- Minimisation of aggregate formation of some IgG isotypes (IgG3 for instance) requires storage at 4°C.
Contamination prevention
Purified custom-made antibodies from Eurogentec already contain 0,01% thimerosal to avoid microbial growth, as mentioned on the data sheet. But please be advised that any serum, like Test Sera for animal selection, pre-immune sera (PPI), large bleed sera (GP), or final bleed sera (SAB) do not contain any preservative.
Please be advised of the following prior to use sodium azide for storage :
- Azide is toxic to organisms and should be avoided in applications which are done with living cells, or tissues (the same is the case for thimerosal – please advise us before starting your immunization programme of your research purpose in order to avoid any preservative);
- If antibodies should be conjugated or spotted involving amino groups, azide will interfere negatively into the process, thimerosal is an acceptable alternative without these unwanted side effects.
Preservatives, if already in an antibody formulation, can be removed in the following ways:
- By dialysis or centrifugal filtration: using a device with a MWCO of 14 kDa, please refer to the manufacturer’s recommendations for most efficient use centrifugal filter devices or dialysis equipment (MW of sodium azide ~ 65 D, thimerosal ~ 404 D, IgG ~ 160 kDa, IgM ~ 600 kDa);
- By gel filtration (which is faster than dialysis). Please refer to the manufacturer’s recommendations for most efficient use of suited filter cartridges
please be advised that all materials used here should be sterile to ensure the subsequent stability of the antibody, and to avoid contamination by purification.
Damage caused by freezing and thawing of antibodies
The antibody’s binding capacity can be reduced by aggregate formation due to denaturation following cycles of freezing and thawing.
Though a lot of researchers keep their antibodies at –80°C, there is no real explanation why –20°C might be inappropriate. In any case, antibodies should rather be stored protected from daily routine in the freezer, e.g. rather in the back than in the front, or in drawers than standing free accessible.
Working concentrations of antibodies should not be stored for more than one day at 4°C, since proteins are more susceptible to degradation if they are stored in lower concentrations. Ideal would be to formulate the antibody with additional protein, like BSA or milk powder to reach concentrations of total protein in the mg/ml range. BSA or milk powder in working solutions also minimizes antibody loss due to attachment to the vessel wall.
The addition of stabilizing proteins is not recommended for antibodies that are subject to conjugation or array spotting.
What information does the ELISA test provide to you ?
The ELISA test carried out by Eurogentec is an indirect ELISA. This means that a constant amount of antigen has been coated into the wells of the ELISA plate (100 ng/well), and tested with different dilutions of the serum or antibody in question. The development is done colorimetric, using a secondary HRP-conjugated antibody, and o-phenylenediamine as chromogenic substrate. The optical density of the chromogenic substrate is measured at 492 nm OD(492).
The method has to be considered as semi-quantitative, since the measured reactivity with growing dilutions can be due to:
- The concentration of specific antibodies against the antigen
- The affinity of the evolved antibodies against the antigen
The results are influenced by the following factors:
- The general success of the immunization against the antigen
- The suitability of the antigen (peptide or protein) to be coated onto ELISA plates
- The exposure of the immunization relevant epitopes after coating
The measured optical densities at 492 nm are plotted against the dilution of the antibody or serum. The following curve types can be found:
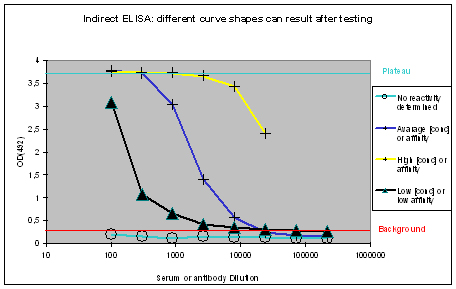
Figure 1 : Different shapes of ELISA curves that can be found after ELISA testing. “No reactivity determined” (light blue), “Low [conc.] or affinity” (black), “Average [conc.] or affinity” (blue), and “High [conc.] or affinity” (yellow) display flat, hyperbolic, or sigmoidal ELISA curve shapes. The optical density OD(492) can be correlated to antibody affinity or concentration in the window between the “Background” (red), and Plateau (turquoise).
Flat ELISA curves
Flat ELISA curves (Figure 1, light blue) are typical for pre-immune sera. Since the immunization host did not encounter the antigen, no immune response could be determined. A flat curve can also result, if antigens coated with the epitope towards the ELISA plate, or which did not coat at all. Potential program difficulties: a flat ELISA curve can result in rare cases, if the antigen is a serum protein, cell surface expressed, and evolutionary conserved – the generation of an immune response will just be suppressed, in order to protect the host (which is normal).
Given all the variables, testing and establishment of the antibody should be carried out accordingly.
Hyperbolic ELISA curves
Hyperbolic ELISA curves (Figure 1, black) are typical for the immune response generation phase during an immunization program. This reactivity kinetics can be found very often when testing the small test bleed (PP) of 87-day programs. The curve might be understood as “a low concentration of high affinity antibodies” has been generated, but it also can result from antigen coating difficulties. Given this, testing the antibody in your application might give completely different results.
Sigmoidal ELISA curves
The first case of sigmoidal ELISA curves represents the average of ELISA tests carried out by Eurogentec. In Figure 1 the curve is represented in blue. The curve falls in a sigmoidal shape from the “Plateau” of saturation usually at a dilution of 1:1.000 towards the “Background”, which is reached with a dilution higher than 1:10.000. The curve might be read like “the immunization provided a mixed population of high and low affine antibodies, which detect the antigen even if highly diluted”. Antibodies or sera producing curves like this can be used in a lot of applications, since the antibody population is rich – the ELISA test only detects antibodies that bind to the epitope immobilised on the ELISA plate, other populations of the antibody are not reflected here.
The second case of sigmoidal ELISA curves is over the average of ELISA tests carried out by Eurogentec. This curve is displayed in yellow in Figure 1. The curve is characterised by keeping the “Plateau” level of saturation very long, and falling in a sigmoidal fashion towards the “Background” level beyond the concentration range considered for ELISA plots. Curves like this result from very immunogenic peptides, large proteins like maybe a provided antigen from customers or the peptide carrier protein KLH used by Eurogentec, or so called “super antigens”. While strong reactivity against your peptide, or protein is desired, and a strong reactivity against KLH is a positive control for general successful immunization using peptides, super antigens represent epitopes that can be found randomly. This random occurrence of structural instead of sequence-related motifs implies as well that the antibody providing this is non-specific. The cases where immunizations provided antibodies to super antigens are rare. Antibodies or sera producing curves like this can usually be used in a lot of applications, since the antibody population is richly composed.
Purifications
After an immunization, you might have included affinity purification of the produced antibodies against the peptide or your antigen in the sufficient amount. Eurogentec offers different formats of purifications, 5 ml, 20 ml and 50 ml. The ELISA can be added optionally to 5 and 20 ml purifications, but it is a part of 50 ml affinity purification. The ELISA has to be considered here, as a mean of characterisation for purified antibodies, and not as a tool providing a quality control. As above already mentioned, the results of the ELISA tests are influenced by
- The accessibility of the epitope for the antibody
- The concentration and plating suitability of the antigen
- Simply the fact that the antibody has been purified with the antigen coupled to a matrix, and is now probed with the antigen immobilised on an ELISA plate (structural differences of the antigen in two different applications)
Given these factors, different results might be possible for the ELISA of the purified antibodies:
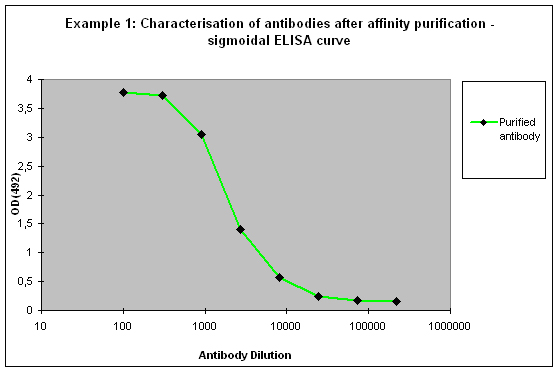
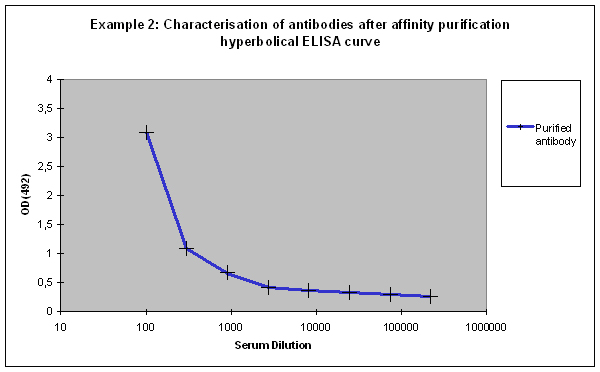
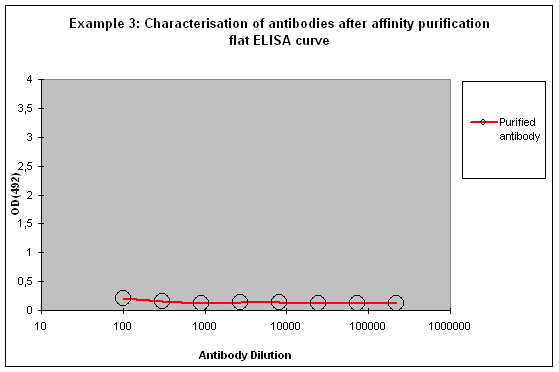
Purification 3 : a flat ELISA curve might suggest that the purification did not provide any working antibody (Example 3). Please be advised that also in such a case testing is worth the effort, since it might still be possible that the antigen looks different on the ELISA plate compared to its structure during affinity purification, and finally in your application. Try to characterise the antibody by Western or dot blotting – these applications work with far over 70% of custom-made antibodies from Eurogentec. A starting dilution might be 1:500. If it tests positive in Western blot – or dot blot, and you want to use your new antibody in your desired application, you have to establish it accordingly, maybe change existing protocols. Please compare the performance of the purified antibody always to the serum (1:500 to 1:10.000 diluted, depending on your optimisation results) in paralleled tests on identical samples. Should you experience difficulties, please feel free to contact us for further advice.
ELISA and Cross Purifications from Phospho-, Methyl-, or other modification specific programs
Since protein modifications play a key role for different cellular functions, and scientific interest for the different types of protein editing events is gaining more attention, Eurogentec is proud to make the detection of post-translational modifications also possible to you.
Immunization programs for their detection can be carried out in rabbits or guinea pig, as Eurogentec’s standard 87-day mode or as 28-day Super Speedy immunizations, but the crucial point is the ELISA testing during, and after the programs.
Identifying the best host for cross affinity purification of modification specific antibodies by ELISA
As in any immunization program carried out by Eurogentec two hosts (in this case rabbits or guinea pigs) have been immunised with a peptide carrying the modified amino acid that you want to detect in down stream experiments.
To identify the best responding host, the pre-immune serum (PPI), and the final bleeds (SAB) of the hosts are tested against the modified peptide and against the carrier protein by indirect ELISA (Figure 2)
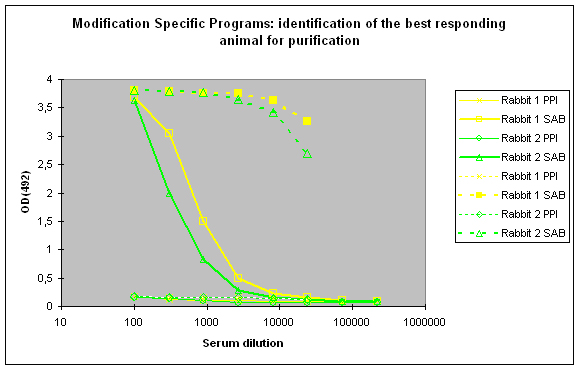
Figure 2 : In this anti-phospho program the immune responses against the phosphorylated peptide (solid lines, rabbit 1, yellow, and rabbit 2, green) and against the carrier protein (dashed lines) are determined by Indirect ELISA (Coated antigen, two hosts, and two bleeds, part of modification specific programs) and compare the reactivity of the serum before immunization (pre-immune serum, PPI, day 0), and the final bleed (SAB, end of the program). The serum to be chosen for cross purification based on Eurogentec’s expertise would be rabbit 1, since the response here is a little bit better compared to rabbit 2 (SAB, solid green and yellow lines). This impression is also supported by the finding that the response from rabbit 1 against the carrier protein (SAB, dashed yellow line) is a little bit better than the carrier response of rabbit 2 (SAB, dashed green line). In analogy to Fig. 1 (above) spoken, both responses to the peptides are good, and similar, but rabbit 1 (SAB, solid yellow line) produced a response a little bit closer to the ideal sigmoidal shaped curve.
Purification 1 : isolation of antibodies that are specific to the peptide and to the modification from rabbit 1
After the best responding host has been identified by the above-performed indirect ELISA, the final bleed serum (SAB) of this host will be used for cross purification. The first affinity purification will be carried out against the modified peptide. By this step the serum will be depleted from antibodies that are specific to the modification, and the peptide context in general. Following this purification, the serum (S), the flow through 1 (FT1), and the purified (PA) are tested by indirect ELISA against the modified peptide and the carrier protein. In general the following results can be expected (Please see also Figure 5) :
|
Fraction |
Reactivity against mod. peptide |
Reactivity against carrier |
|
Purified antibody (PA) |
Yes, stronger than S, and much stronger than FT1 |
No, if Yes then removal by dilution quickly possible |
|
Flow through 1 (FT1) |
No, if Yes than much weaker than S or PA |
Yes |
|
Serum (S) |
Yes, weaker than PA |
Yes |
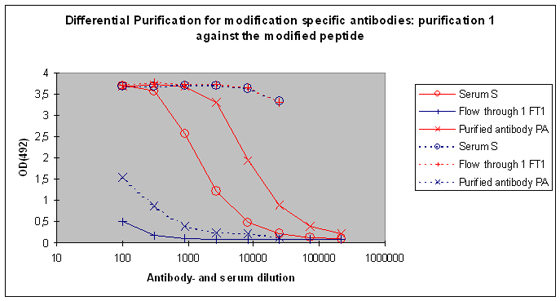
Purification 2 : separation of modification specific antibodies from just peptide specific antibodies
Peak 2 and FT2 will be characterised by indirect ELISA against the modified peptide, and against the non-modified peptide. In general the following results can be expected:
|
Fraction |
Reactivity against mod. peptide |
Reactivity against the non-modified peptide |
|
Peak 2 (P2) |
Yes, the modified peptide contains a lot of antigens |
Yes, the modified peptide contains a lot of antigens |
|
Flow through 2 (FT2) |
Yes |
No, if any it can easily be diluted away |
A sample result is reflected in Figure 4 and Figure 5.
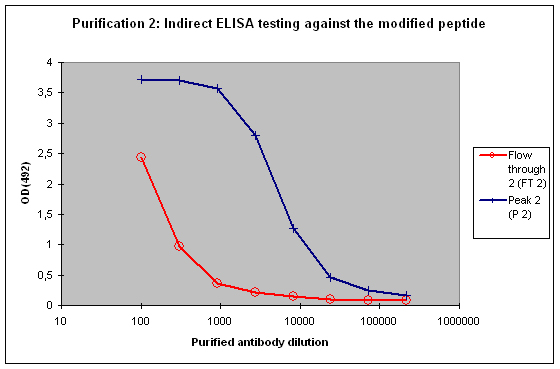
Figure 4 : Testing of Peak 2 (P2) and flow through 2 (FT2) against the modified peptide by indirect ELISA. Antibodies separated with the non-modified peptide react also with the modified peptide – these antibodies cannot be considered as a control for the non-modified protein target in your experimentation. The flow through 2 (FT2) antibodies show a weak reactivity against the modified peptide – please consider that the modification in the peptide might directly interact with the ELISA plate surface, and therefore a restricted access of antibodies to the modified epitope might result, causing a hyperbolical curve shape.
Please be advised that the population of antibodies against the modification in the deciding fraction FT2 can be very low, due to narrowing the variety of specific molecules towards a single feature of a small epitope. But nonetheless, the antibodies can be used in the most common application types diluted in the same way like other polyclonal antibodies.
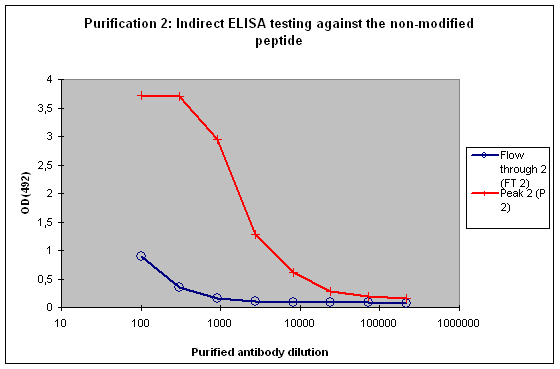
Figure 5 : Testing of Peak 2 (P2) and flow through 2 (FT2) against the non-modified peptide by indirect ELISA. P2 contains the majority of antibodies to the non-modified peptide, which is reflected by the strong reactivity of the antibodies to the target. FT2 might contain a small population also specific for the non-modified peptide. These antibodies can be diluted out or washed off in down stream applications, the soon drop of the curve from FT2 indicates that these antibodies are no good binders.
Please contact us if you wish to discuss your ELISA results in detail, or if you wish support for the establishment of your antibody.
Antibody Evolution (meaningful for 87-day immunization)
Eurogentec offers you the possibility to have ELISA testing done – this can be chosen as option for any immunization program. Simply ask for a complete program including ELISA right at the start (Please note, ELISA testing represents for 28-day Super Speedy immunization a set of supplementary characterisation data – the test can for technical reasons not be performed to decide on programme extension or stop).
In case ELISA testing with your program is ordered, we carry out the same standard program however without sending the peptide and sera directly to the customer. We keep the free peptide, the preimmune and small test sample (obtained after 3 injections) until the large bleed sample is available (day 66, after the 4th injection). At this moment, we carry out the ELISA test using one 96-well micro titre plate per animal. We test in parallel per ELISA dilutions from:
- Preimmune (PPI),
- Small (PP) and
- Large (GP) test bleed
Against
- The free peptide and
- Against the carrier protein,
- Including positive and negative controls.
The test report is sent to you per e-mail, and this is as well announced in the program related immunization schedule.
This paralleled testing not only allows to have a good view of the antibody evolution with the last boost, but it also gives a relatively good decision facilitation for program prolongation with additional boosts and bleeds, termination as scheduled, or immediate termination (Example 1).
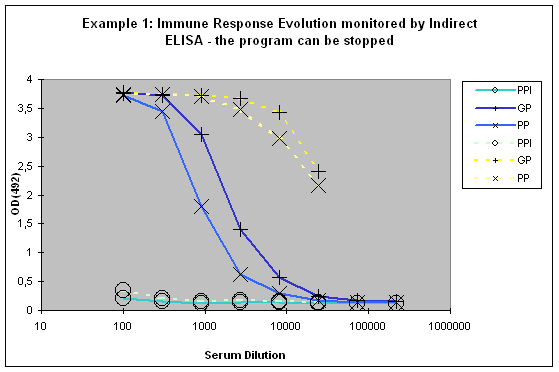
Figure 6 : In this anti-peptide program the immune responses against the peptide (solid lines) and against the carrier protein (dashed lines) are determined by Indirect ELISA (Coated antigen, one host, and three bleeds, referenced as AS-ELIS-01) and compare the reactivity of the serum before immunization (pre-immune serum, PPI, day 0), the small test bleed (PP, day 38), and the large bleed (GP, day 66). The success of the program is reflected by the sigmoidal curve shape of the plot, which evolved from “no response” (PPI), already strong responses after day 38 (PP), and the strongest responses after day 66 (GP). The program in this host can be finished, if desired, since the response evolution reached already at day 66 a maximum being characteristic for day 87 (the end point of a conventional 87-day program).
If a good increase is observed between the first and the second test bleed, the chances to get still higher titres with additional injections are quite good (Example 2). If however both bleeds (first and second test bleed) show more or less the same titre, an additional injection will most of the time result in only slight or no titre increase of specific antibodies.
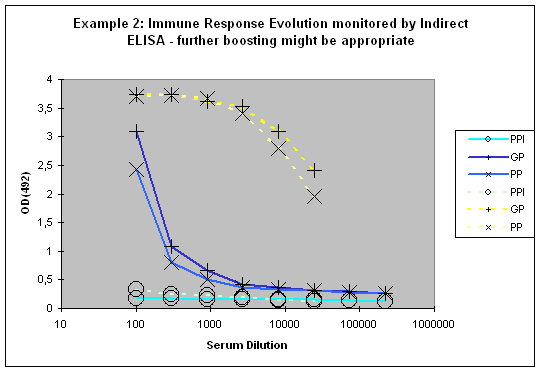
Figure 7 : In this anti-peptide program the immune responses against the peptide (solid lines) and against the carrier protein (dashed lines) are determined by Indirect ELISA (Coated antigen, one host, and three bleeds, referenced as AS-ELIS-01) and compare the reactivity of the serum before immunization (pre-immune serum, PPI, day 0), the small test bleed (PP, day 38), and the large bleed (GP, day 66). The antibody evolution of the program is slow, and reflected by the hyperbolic curve shape for the PP and GP for the peptide (blue) of the plots. The carrier (yellow, compare also to Fig. 1) produced already strong responses, which evolved from “no response” (PPI), to almost maximum at day 38 (PP), and the strongest responses after day 66 (GP). The program in this host should be extended by additional boosts in order to receive a sigmoidal curve shape (compare Fig.: 1, PP and GP, blue) reflecting the desired antibody reactivity against the antigen (Compare to blue curves PP and GP in Fig.: 1).
Please be advised that ELISA results are telling the customers and us if the immunization worked. Nonetheless, this information can also bear weaknesses. Usually proteins bind very good to ELISA plates but especially small antigens like peptides or other haptens might
- Not bind at all (a false negative result)
- Bind insufficiently (the result might partially reflect the immune response which has been evolved in the animal)
- Bind to the plate with by the epitope (a false negative result)
Please note the general efficiency in case of a peptide immunization program is reflected by the strong immune response to the carrier protein (a global positive control, Fig.: 1 and 2, yellow curves). If the response against the peptide appears to be strikingly weaker (Fig.: 2 for example; Fig.: 1 reflects rather the ideal case), you should in each case test the serum in your intended application, or by a different method, like dot or Western blotting.
Please note, it is hardly possible to extrapolate results from working ELISA tests to other applications, or to conclude from an antibody’s efficiency in one application to its potential capabilities in other applications: this means that an antibody providing bad results in ELISA testing, might be excellent in your down stream application, where the antigen looks different – but also vice versa is possible.
Please contact us if you further wish to discuss your ELISA results.
General considerations:
Please be advised that the antigen binding characteristics of an antibody after affinity purification might be shifted from polyclonal to a monoclonal-like specificity. A protein as antigen has much more epitopes for antibodies than a peptide. It is therefore in a few instances likely that especially in the case of peptides that key epitopes get hidden on ELISA plates, and ELISA results do not reflect properly the detection capabilities of the tested antibody, like they might be revealed in your intended application.
Eurogentec works with standardised protocols for purification of antibodies. Though these protocols are optimised, it might still be possible in rare cases that antibodies are not completely eluted from the purification column, nor can it be excluded that all partially denatured antibodies fold back to functional molecules during neutralisation after elution. In these cases affinity purification might not be the right way to concentrate your antibody.
Please note that immunization hosts are not transparent, and very rare occurring failure of immunization programs are not foreseeable to us, Eurogentec cannot guaranty for any antibody efficiency, nor warrant any quantities or capabilities of the produced molecules.
Which antigen formats are compatible with an immunization?
Lyophilized
You can send us your freeze-dried antigen at ambient temperature. If the antigen is poorly soluble in aqueous solution, we suggest that you avoid lyophilization and that you send us your protein in solution on dry ice. The addition of adjuvant will help to dissolve even lipophilic proteins. If the antigen does not dissolve, a fine suspension of the antigenwill be obtained by thorough mixing. Such fine suspensions can also be useful for antibody production because particles are efficiently phagocyted.
SDS-PAGE**
For antibody productions with SDS-PAGE gel fragments, we advise our customers to cut out the band of interest and to aliquot it in separate tubes for each injection. The antigen tubes can be shipped at room temperature. The standardCoomassie and Coomassie-like staining procedure can be used since the Coomassie staining dyes do not interfere with the antibody evolution. However, sylver stain is not allowed.
The band should just be washed briefly but thoroughly in water to remove acetic acid and methanol residues, and then cut into injection pieces, and aliquoted wet into safe lock tubes to avoid drying. The gel must not be dried or lyophilized, because this would make the fragmentation before injection more difficult.
** Not available for Speedy programme or small rodents.
In Solution
You can send us your antigen in solution on dry ice. We recommend limiting as far as possible the use of detergents and aggressive chemicals such as acetic acid, guanidine hydrochloride, heavy metals and other agents that are toxicto the host animal.
It is possible to immunize animals with an antigen solution containing 8M-urea, but this is more painful for the rabbits.For this reason, we ask our customers to send us the antigen as concentrated as possible so that we can dilute the solution before injection in order to decrease the final urea concentration. Antigens in solution should be sent in avolume not exceeding the authorized amount per injection.
e.g.:
- Rabbit 500 µL/injection
- Rat 250 µL/injection
- Guinea Pig 250 µL/injection
- Mouse 150 µL/injection
- Hen 500 µL/injection
- Goat/Sheep 100 µL/injection.
How should I store my serum?
Storage Suggestion for Antibodies and Serum used for in vitro Research Applications
Properly stored, antibodies should remain stable for months or years. Freezers with automatic de-frosting are absolutely inappropriate for antibody and serum storage.
If you are using catalogue antibodies, please refer to the provided data sheet.
Storage Temperatures
Our purified antibodies are delivered to you in PBS with 0,1% BSA as stabiliser and 0,01% Thimerosal as preservative. For these formulations we suppose that storage of small aliquots at –20°C is appropriate.
The aliquoting of antibodies has the following advantages:
- Minimisation of the damage due to freezing and thawing
- Exclusion of contamination due to multiple pipetting from a single vial
Aliquots should be thawed once, with any remaining antibody kept at 4°C.
When you receive an antibody you should carefully thaw it (in the fridge in a 4°C water bath – do not forget about it) and centrifuge it with 10.000 x g for 20 sec in order to pull down all the solution. Aliquot the antibody in low-protein-binding micro centrifuge tubes for storage. Please adapt the size of the aliquots to your typical experimental consumption. Aliquots should not be smaller than 10 µl to avoid damage of the antibody by evaporation and adhesion to the vessel wall.
Though it is considered in general as optional, we recommend the addition of 50% glycerol (v/v, final) to your antibodies for additional freezing and evaporation protection for storage at –20°C (glycerol is frozen at –80°C).
Please use just sterile chemicals and solutions for your antibodies to prevent down stream microbial growth.
Upon receipt, antibody storage for one or two weeks (time required for experimental establishment) at 4°C should be acceptable with subsequent aliquoted freezing for long-term storage.
Ascites and serum represent an exception here since the contained proteases require immediate storage at -20°C or -80°C.
Please be advised that sera produced at Eurogentec do not contain any preservative, 0,01% azide or thimerosal (depending on your research purpose) should be added to the aliquots for storage at –20°C after first thawing.
Egg yolks in contrast must never be frozen, since they denature into a form that does not allow isolation of IgY and affinity purification anymore. Egg Yolks from Eurogentec are shipped at 4°C with 0,01% azide added as preservative ready for short-term storage (over several weeks) at 4°C.
For long-term storage we recommend the purification of IgY from the egg yolks.
IgY isolates and affinity purified IgY antibodies should be treated like purified antibodies described in the beginning of this section.
Antibody formulations that require special attention:
- Enzyme-conjugated antibodies should not be frozen at all in order to retain the maximum enzymatic activity, storage at 4°C is therefore mandatory, please refer to the technical datasheet on the solvent formulation, and add if required 0,01% Thimerosal (Azide will inactivate HRP-conjugates)
- All conjugated antibodies (enzymes, fluorophores, or biotin) should be stored in the dark (very well suited: the black foil bags of x-ray films or photographic paper, and the corresponding boxing)
- Fluorophore-conjugated antibodies can be stored aliquoted at –20°C with 50% glycerol (final v/v) – please make sure by the data sheet that they are formulated as well with BSA, physiologically concentrated buffer salts (e.g. PBS), and preservatives (azide or thimerosal), furthermore, please be sure to use a sterile glycerol formulation, since microbial growth might occur and destroy the antibody. Storage at –80°C of glycerol containing solutions is not advised since this is below the freezing point of glycerol
- Minimisation of aggregate formation of some IgG isotypes (IgG3 for instance) requires storage at 4°C
Contamination prevention
Purified custom-made antibodies from Eurogentec already contain 0,01% thimerosal to avoid microbial growth, as mentioned on the data sheet. But please be advised that any serum, like Test Sera for animal selection, pre-immune sera (PPI), large bleed sera (GP), or final bleed sera (SAB) do not contain any preservative.
Please be advised of the following prior to use sodium azide for storage
- Azide is toxic to organisms and should be avoided in applications which are done with living cells, or tissues (the same is the case for thimerosal – please advise us before starting your immunization programme of your research purpose in order to avoid any preservative)
- If antibodies should be conjugated or spotted involving amino groups, azide will interfere negatively into the process, thimerosal is an acceptable alternative without these unwanted side effects
Preservatives, if already in an antibody formulation, can be removed in the following ways:
- By dialysis or centrifugal filtration: using a device with a MWCO of 14 kDa, please refer to the manufacturer’s recommendations for most efficient use centrifugal filter devices or dialysis equipment (MW of sodium azide ~ 65 D, thimerosal ~ 404 D, IgG ~ 160 kDa, IgM ~ 600 kDa).
- By gel filtration (which is faster than dialysis). Please refer to the manufacturer’s recommendations for most efficient use of suited filter cartridges
please be advised that all materials used here should be sterile to ensure the subsequent stability of the antibody, and to avoid contamination by purification
Damage caused by freezing and thawing of antibodies
The antibody’s binding capacity can be reduced by aggregate formation due to denaturation following cycles of freezing and thawing.
Though a lot of researchers keep their antibodies at –80°C, there is no real explanation why –20°C might be inappropriate. In any case, antibodies should rather be stored protected from daily routine in the freezer, e.g. rather in the back than in the front, or in drawers than standing free accessible.
Working concentrations of antibodies should not be stored for more than one day at 4°C, since proteins are more susceptible to degradation if they are stored in lower concentrations. Ideal would be to formulate the antibody with additional protein, like BSA or milk powder to reach concentrations of total protein in the mg/ml range. BSA or milk powder in working solutions also minimizes antibody loss due to attachment to the vessel wall.
The addition of stabilizing proteins is not recommended for antibodies that are subject to conjugation or array spotting.
What are the authorized buffer components for immunization?
Compound or formulations
| Buffer | Allowed | Not allowed |
| Water | Yes, keep the volume small (1 ml) | - |
| PBS | Yes, keep the volume small (1 ml) | - |
| Physiological buffer solutions |
Yes, keep the volume small (1 ml) | - |
| Metal dyes/heavy metals | - | Risk of toxicity |
| Salts (KCl, NaCl, MgCl2 | < 1.0 M | > 1.0 M |
| SDS | < 2.0 % | > 2.0 % |
| Urea | < 6.0 M | > 8.0 M |
| Guanidinium HCl | - |
Risk of toxicity |
| Digoxin/Digoxigenin | - | Risk of toxicity |
| Octylglucoside | <1.0 % | > 1.0 % |
| Triton X-100/Tween-20 | < 0.2 % | > 0.2 % |
| Glycerol | < 20 % | > 20 % |
| PMSF | - | Risk of toxicity |
| Pefabloc | < 0.1 mM | > 0.1 mM |
| Leupeptin/Pepstatin | < 1 μM | > 1 μM |
| DTT | < 3 M | > 3 M |
| Mercaptoethanol | - | Risk of toxicity |
| Imidazole | < 3 M | > 3 M |
| TFA | - | High risk of toxicity |
How to provide my antigen?
Lyophilized
You can send us your freeze-dried antigen at ambient temperature. If the antigen is poorly soluble in aqueous solution, we suggest that you avoid lyophilization and that you send us your protein in solution on dry ice. The addition of adjuvant will help to dissolve even lipophilic proteins. If the antigen does not dissolve, a fine suspension of the antigen will be obtained by thorough mixing. Such fine suspensions can also be useful for antibody production because particles are efficiently phagocyted.
SDS-PAGE**
For antibody productions with SDS-PAGE gel fragments, we advise our customers to cut out the band of interest and to aliquot it in separate tubes for each injection. The antigen tubes can be shipped at room temperature. The standard Coomassie and Coomassie-like staining procedure can be used since the Coomassie staining dyes do not interfere with the antibody evolution. However, Sylver stain is not allowed. The band should just be washed briefly but thoroughly in water to remove acetic acid and methanol residues, and then cut into injection pieces, and aliquoted wet into safe lock tubes to avoid drying. The gel must not be dried or lyophilized, because this would make the fragmentation before injection more difficult.
** Not available for Speedy program or small rodents.
In solution
You can send us your antigen in solution on dry ice. We recommend limiting as far as possible the use of detergents and aggressive chemicals such as acetic acid, guanidine hydrochloride, heavy metals and other agents that are toxic to the host animal. It is possible to immunize animals with an antigen solution containing 8M-urea, but this is more painful for the rabbits. For this reason, we ask our customers to send us the antigen as concentrated as possible so that we can dilute the solution before injection in order to decrease the final urea concentration. Antigens in solution should be sent in a volume not exceeding the authorized amount per injection.
For ex:
- Rabbit 500 µL/injection
- Rat 250 µL/injection
- Guinea Pig 250 µL/injection
- Mouse 150 µL/injection
- Hen 500 µL/injection
- Goat/Sheep 100 µL/injection
Labeling and Dyes
What is the recommended protein concentration when using an AnaTag labeling kit?
A concentration of ≥ 2mg/mL is recommended.
In which buffer can my protein be dissolved in when using AnaTag kits?
See the product data sheet for recommended buffers. It is important to avoid buffer additive reducing agents (e.g. DTT), protein stabilizers (e.g. BSA) or sodium azide. Additionally, the following buffers should be avoided: tris, glycine, ammonium salts and buffers containing free amines.
How can free dyes be removed from my labeled protein?
The free dyes are removed during the purification step via the spin column that is provided with the kit.
What purification yields can I expect?
On average, we observe 70-80% yields.
After labeling my protein, can I use a dialysis membrane to remove excess dye?
Yes, but it is important to use a membrane molecular weight cut off that is small enough to prevent protein from being lost through the membrane pores.
Where does the binding occur between the dye and my protein?
Dyes in AnaTag kits have a succinimidyl ester reactive group which can bind to any free amine available on your protein. Hence the dye can bind to any Lysine residues and protein N-terminus.
Nucleic Acid Electrophoresis
Do the SmartViewer can be used on any electrophoresis device ?
No, it doesn’t, The SMARTViewer is designed to fit the Mupid® device for nucleic acid electrophoresis.
Nucleic acids extraction & purification
What samples can be used with the SmartExtract DNA Extraction kit ?
The SmartExtract DNA Extraction kit achieves genomic DNA extraction from almost any crude sample type (e.g. blood, human & animal tissues, plant seeds & leaves…).
How long does it takes to recover DNA with the SmartExtract DNA Extraction kit ?
DNA extraction is performed in a single tube in 12 minutes only.
What samples can be used with the SmartPURE DNA Purification kits ?
There are 3 kits available according to the sample source.
We recommend to use the SmartPure Gel Kit for DNA purification from standard high or low melting agarose gel.
For DNA purification from PCR mixes, the SmartPure PCR Kit is indicated.
For plasmid Mini-purification from bacterial cell culture, please use the SmartPure Plasmid Kit.
How long does it takes to recover DNA from Agarose Gel with the SmartPURE Gel Kit ?
DNA purification from agarose gel takes on average 16 min.
How long does it takes to recover DNA from PCR Mixes with the SmartPURE PCR Kit ?
DNA purification from PCR Mixes takes on average 6 min.
How long does it takes to isolate and purify Plasmid from bacterial cell culture with the SmartPURE Plasmid Kit ?
Purified plasmid can be collected in 20 min on average.
What is the typical yield of plasmid recovery with the SmartPURE Plasmid Kit?
Yield may vary according to the plasmid copy number and the culture OD but the typical yield obtained is up to 40 µg plasmid DNA/1-5 mL E.coli culture.
What is the DNA size range covered by the SmartPURE Gel Kit ?
The SmartPURE Gel Kit can perform extraction and purification of DNA sequence from 60 bp to 23kbp.
What is the DNA size range covered by the SmartPURE PCR Kit ?
The SmartPURE PCR Kit can perform extraction and purification of DNA sequence from 60 bp to 10kbp.
Oligonucleotides
How do I reconstitute my oligos?
- Spin the tube briefly to collect the pellet in the bottom of the tube.
- Add an appropriate volume of sterile TE buffer (10 mM Tris-HCl, 0.1-1 mM EDTA ; pH 7.5-8.0) or dH2O.
- Allow the tube to stand for a few minutes at room temperature.
- Stir the tube for 15 seconds using a vortex shaker and spin it briefly.
Stock solution
We recommend preparing a stock solution at 100 µM concentration which can be achieved by adding a volume (µl) of sterile TE buffer or dH2O, equal to ten times the number of nanomoles of sample present in the tube.
Florescent dyes
For optimal long-term storage of fluorescent dye-labeled oligonucleotides, it is recommended that the oligonucleotides be resuspended in a slightly basic solution (i.e., sterile TE buffer at pH 8).
Exception: Cy® dye-labelled oligonucleotides should be resuspended at pH 7.
siRNAs
(si)RNA should be resuspended in RNase-free buffer to a convenient stock concentration (20 to 50 µM) and in small aliquots to avoid multiple freeze thaw cycles and contact with RNases.
Difficult resuspension
Please note that some oligonucleotides (i.e. milligram amounts or phosphorothioate oligonucleotides) are particularly difficult to resuspend and may require longer incubation times and/or thorough vortexing.
Heating may also help to speed up the process.
What does synthesis scale mean?
The synthesis scale refers to the amount of raw material used to start the synthesis of oligonucleotides.
The yield corresponds to the amount of final product recovered at the end of the synthesis and purification processes.
Which factors do influence the oligonucleotide production yield?
The oligonucleotides production yeld may be influenced by the following factors:
- The presence of one or multiple modifications
- The nature of the modifications
- The coupling method and efficiency
- The purification method
How to calculate the oligo concentration from absorbance at 260 nm?
To quantify your Oligonucleotides, make an aliquot of the resuspended Oligonucleotides to a final volume of 1 ml of dH2O and vortex for a few seconds.
Measure the absorbance of this dilution at 260 nm (A260). Use the formula below to calculate the concentration of Oligonucleotides in your stock solution.
This formula is valid for an absorption of A260 ≤1.2.
Concentration in µg/ml = A260 x dilution factor x Weight per OD of stock solution (in µg/OD)
How Optical Density (O.D.) at 260 nm is linked to oligo quantity?
1 OD260 (Optical Density) unit is defined as the amount of oligonucleotide which, when dissolved in a volume of 1.0 ml results in an absorbance of 1.0 when measured at 260 nm in a 1 cm path-length quartz cuvette.
1 OD260 unit corresponds to approximately 33 μg of single strand DNA. These relationships, however, can be inaccurate for short fragments of DNA, such as Oligonucleotides. Base composition and even linear sequence will affect optical absorbance. Hence the precise value of the OD to mass relationship is unique for each oligonucleotide.
Example:
1.0 OD260 of CCCCCCCCCC (10 bases) equals 39 μg
whereas 1.0 OD260 of AAAAAAAAAA (10 bases) equals only 20 μg
How to convert OD260 into nanomoles?
The following equation shows the relation between the oligo amount in nanomoles and the OD 260 value
Nanomoles = (OD260/ε260)x106
ε260 is the extinction coefficient at 260 nm
Example:
1 OD260 unit of primer M13 Forward,
5’-GTA AAA CGA CGG CCA GTG-3’
Molar extinction coefficient (ε260) = 182.800 L / (mole x cm)
Nanomoles = (1.0 / 182.800 ) × 106 = 5.47 nmoles
How to convert nanomoles into microgrammes?
Using the following equation, it is quite simple to calculate the amount in microgramme from the nanomole value and the Molecular weight of the oligonucleotide.
Micrograms = Molecular Weight × Nanomoles × 10-3
Example:
1 OD260 unit of primer M13 Forward,
5’-GTA AAA CGA CGG CCA GTG-3’
Molecular Weight = 5558.7
Micrograms = 5558.7 × 5.47 × 10-3 = 30.4 μg
How to calculate the molar extinction coefficient?
Molar Extinction coefficient can be calculate by the following formula
ε260=2x (∑1(n-1)εNearest Neighbour) - ∑2(n-1)εindividual + ∑1nεModification
where ΣNearest Neighbour is the nearest neighbour constant for a pair of bases, ΣIndividual is the constant for an individual base, and n is the length of the oligonucleotide.
How to calculate the molecular weight of my oligonucleotide?
Anhydrous MW (g.mol-1) = ∑individual base MW + ∑individual Modification MW - 63.98 + 2.016
For DNA bases: MW dA = 313.21; MW dC = 289.18; MW dG= 329.21; MW dT = 304.20; MW dU= 290.17; MW dI = 314.19
For RNA bases: MW DNA counterpart + 16. When determining the weight of Uracil (rU) start with dU and not dT
For LNA bases: MW dA = 313.21; MW dC = 289.18; MW dG = 329.21; MW dT = 304.20; MW dU = 290.17; MW dI = 314.19
For 2’ O-Methyl bases: MW DNA counterpart + 30.03. When determining the weight of mU start with dU and not dT
For phosphorothioated bases: MW DNA counterpart + 16.06
How long do my oligos remain stable?
Our Quality Control department performs regularly stability studies on different type of oligonucleotides. Each oligo is stored under different conditions and results are analyzed to provide to our customers the most accurate information regarding the stability of their products.
How are Eurogentec's oligos made and which chemistry is used?
Oligonucleotides are produced on nucleic acid synthesizers using optimized phosphoramidite chemistry and fully automatic oligo synthesis.
Our proprietary synthesis platforms provide computer-controlled oligo synthesis, cleavage, and deprotection in 4 different processing formats.
Even though oligos are synthesized with the highest achievable coupling efficiencies, we always perform an additional purification step on every oligo batch.
This purification step ensures that even the simplest primer will be suitable for most molecular biology applications, such as PCR, RT-PCR, sequencing, and hybridization studies.
During DNA synthesis, each nucleotide is coupled sequentially (from 3' to 5') to the growing chain according to the standard b-cyanoethyl chemical reactions. Each cycle consists of:
- Deblocking: the first nucleotide, attached to the solid support is deprotected by removing the DMT-protecting group. This produces a free 5' hydroxyl group to react with the next nucleotide.
- Coupling: the next nucleotide is added to the reaction and is covalently attached (i.e. coupled) to the previous nucleotide.
- Capping: any of the first nucleotide that failed to react is capped so that it will no longer participate at any subsequent steps.
- Oxidation: the bond between the first nucleotide and the successfully coupled second nucleotide is oxidized to stabilize the growing chain.
- Deblocking: the 5' DMT group is removed from the second nucleotide to prepare it for further cycles.
At the end of the oligo synthesis, the crude product is cleaved from the solid support (CPG or polystyrene beads) and purified using various methods.
Does my oligonucleotide have a phosphate on the 5' or the 3' end?
All of our custom oligonucleotides are synthesized with a hydroxyl group on both the 3' and the 5' ends.
However, if requested, we can synthesize your oligo with a 5' and/or 3' phosphate.
What is the longest oligonucleotide you can make?
Coupling efficiency in oligonucleotide synthesis is greater than 99 percent and it is possible to synthesize a 200 mer oligonucleotide by special methods developed in the Eurogentec laboratory. We have already succeeded in synthesizing a 220 mer oligo.
However, even though we can produce long oligonucleotides with excellent purity in terms of length, there is another factor to consider. The many chemical steps during each cycle of oligonucleotide synthesis have a small probability of causing damage to the bases (base modifications). Some of these modifications are mutagenic and will result in a product with incorrect coding properties. Such a strand could be isolated during cloning. The base modifications are difficult to remove by standard purification methods used for long oligos and so the best way to avoid them is to be less ambitious about the length of the oligonucleotides you use. Moreover, yield and quality are affected by base composition.
For most practical purposes it is better to synthesize two 100 mer oligonucleotides and ligate them together using a short complementary oligonucleotide template to hold the two long oligonucleotides together for the enzymic ligation step. If necessary one or both of the long oligonucleotides can be chemically phosphorylated.
G-rich oligonucleotides, can they be reliably synthesized?
Special precautions must be taken when synthesizing G-rich oligonucleotides but this is not a problem for us.
Equally, care must be taken when using them in a biological application as they tend to aggregate and form tetrameric structures. This leads to insolubility. Formation of such aggregate can also hinder the ability of the oligo to hybridize to its complementary strand or target sequence.
To avoid this, G-rich oligonucleotides should be dissolved in buffer, heated to just below boiling point then very slowly cooled down in the presence of the complementary strand or target sequence.
Moreover, to avoid long stretch of G, Inosine (a universal base) can be substituted for some of the "g" residues to disrupt the tetraplex.
If you encounter problems using a G-rich oligonucleotide please contact us for further advice.
How do I calculate the melting temperature (Tm) of an oligonucleotide?
The melting temperature (Tm value) of an oligonucleotide is dependent upon the length of the sequence, the G+C content and the type and concentrations of cation present, particularly sodium ion, Na+. We are using the following formulae to calculate the Tm:
Recommended for primers from 14 to 20 bases :
Tm1 (°C) = 2 * (A+T) + 4 * (G+C) (Wallace-Ikatura formula)
Recommended for primers > 20 bases :
Tm2 (°C) = 81.5 + 16.6 * log10[0.05] + 0.41 * (%G + %C) - 675 / N = (81.5 - 21.597098) + 0.41 * (%G + %C) - 675 / N
where N is the length of the oligo. The formula we use takes into account the salts concentration of the reaction, as PCR is typically performed in the presence of ~ 50 mM monovalent cations (0.05 in the above formula).
For degenerated oligos, the lowest (%G + %C) value must be used. For oligos containing Inosine, length = (Length of the oligo) - (Number of Inosine bases). Tm calculation is inaccurate for LNA, PNA and may be inaccurate for oligos containing certain modified bases.
What are the IUB codes for mixed bases?
A=Adenosine
B=[G,T,C]
C=Cytosine
D=[G,A,T]
G=Guanosine
H=[A,T,C]
K=[G,T]
M=[A,C]
N=[A,T,G,C]
R=[A,G]
S=[G,C]
T=Thymidine
V=[G,A,C]
W=[A,T]
Y=[C,T]
How does Eurogentec quantify its oligos?
We carefully measure the OD (Optical Density) value for your custom oligonucleotide by measuring the absorption at 260 nm using an UV spectrometer. This information is provided on the oligonucleotide Technical Data Sheet as: the number of OD260 units, the number of nmoles and the number of µg. The amount of oligo expressed in nmoles and µg is calculated from the OD measurement.
To estimate how much of a freeze-dried oligonucleotide I have should I rely on the weight in mgs or the absorbance (O.D. value)?
When we supply oligonucleotides on a large scale as freeze dried solids we weigh them and also measure the O.D. value as a solution in 1.5ml of water. The O.D. value is used to calculate the number of micromoles and micrograms of DNA. We do not use the weight in mgs for these calculations for the following reason:
A freeze-dried sample of DNA is highly hydrated as water binds very tightly to the major and minor grooves of DNA duplexes and to the edges of the bases, sugars and phosphates of single-stranded nucleic acids. Therefore when the freeze-dried pellet is weighed, the total weight cound be up to 50% water. So why do we weigh the sample? The answer is to make sure that the weight roughly corresponds to the O.D. value. In general (very roughly), 20 O.D. units of mixed sequence DNA will weigh 1 mg. If the weight is much too high relative to the O.D. value we would know that something is wrong. The usual cause for a discrepancy occurs with very short oligonucleotides. They are quite difficult to desalt after HPLC and therefore a high weight relative to the O.D. reading is indicative of contamination with salt. In such a case we would repeat the gel-filtration step to remove traces of salt, re-weigh the sample and re-determine the O.D. value.
What synthesis scale should I order on and what yields do you guarantee?
Please refer to the our Minimum guaranteed amounts
What quality control do you carry out on oligonucleotides?
Quality control (QC) is an indissociable part of any oligonucleotides synthesis process. Eurogentec Oligonucleotides are made using only the highest quality equipment and reagents to guarantee excellent results. All reagents used for oligo synthesis come from reliable suppliers, and each lot is extensively QC checked prior to use.
All our synthesizers are fitted with on-line (real-time) trityl analysis to ensure that synthesis of each oligo meets our stringent quality requirements. All oligos are routinely analyzed by optical density (OD260) measurement.
According to the oligo specifications, we perform quality controls for free as reported in the table below.
|
Oligo Type |
MS1 | UHPLC | |
| Custom Oligonucleotides | Unmodified | ✓2 | |
| Modified | ✓3 | ||
| qPCR Probes | Double Dye Probes | ✓ | ✓ |
| Molecular Beacons | ✓ | ✓ | |
| MGB Probes | ✓ | ✓ | |
| RNAi Oligonucleotides | siRNA Duplexes | ✓ | |
| NGS Oligonucleotides | ✓ | ✓ | |
| Complex Oligonucleotides | ✓ | ✓4 | |
| Calibration Oligos | ✓ | ✓ | |
| Universal Primers | ✓ | ✓ |
MS: Mass Spectrometry; UHPLC: Ultra Performance Liquid Chromatography.
1Always provided up to 60 bases long Oligonucleotides.
2 Randomized high throughput control.
3Except for SePOP desalted oligonucleotides.
4Optional.
For technical reasons this general rule may be adapted to provide you with the most suitable accurate oligonucleotide.
If requested, we can perform specific QC
What is trityl monitoring?
At the end of each coupling cycle, the 5' end deprotecting group: the trityl also called DMT, is removed from the added nucleotide. The trityl quantity is then measured and plotted in a paragraph. Since the chemistry that all oligo manufacturers use does not achieve a 100% coupling, the coupling efficiency is used to predict the quality of the oligo. At Eurogentec we demand a very high coupling efficiency before an oligo will pass our QC.
Is HPLC purification better than cartridge purification?
HPLC purification produces higher yields of purified oligonucleotides than cartridge purification. It gives additional feedback which simple cartridge purification cannot do by providing our skilled technicians with a detailed chromatagram which reveals the purity of the oligonucleotide and indicates if there are any problems which were not picked up in the trityl analysis (synthesis quality analysis). Some impurities in modified oligonucleotides are not removed by cartridge purification (e.g. a common impurity in HEX-labelled oligonucleotides).
How modifications are incorporated into an oligonucleotide?
Oligonucleotides can be modified by direct incorporation during the synthesis or by post-synthesis labeling.
Direct incorporation
3’ modifications
Since automated oligonucleotide synthesis is realized from 3’ to 5’, these modifications are only possible if the corresponding solid support (CPG column) is available and if the modification is compatible with the chemistries used during the synthesis.
Typicalexamples are 3’-phosphate, 3’ Biotin, 3’ FAM, 3’ DDQ I, 3’ BHQ-1®…
5’ and internal modifications
Many modifications can be directly introduced at the 5’ end or at internal positions of the oligonucleotides using the phosphoramidites. However these modifications need to support the somewhat harsh cleavage-deprotection conditions including a strong basic pH.
Typical examples are 5’ Biotin, 5’ Phosphate, 5’ Cholesterol, 5’ FAM, 8-Oxo-dA, Biotin-dT, DABCYL-dT…
Post-synthesis incorporation
Post-synthesis modifications may influence the yield of the reaction. A lower yield may result from poly-modifications and/or strong secondary structures. Two major post-synthesis reactions are used to introduce sensitive dyes or compounds that do not exist as phosphoramidites.
In the first case the label is conjugated to an amino-modified oligonucleotide (3’, 5’ or on a dT) using its amino-reactive version (N-hydroxysuccinimide (NHS) ester in most cases).
The second possibility (originally also used for synthesis of molecular beacons) is the addition of a maleimide-modified label to a thiol-modified oligonucleotide.
Can I have different sugar / backbone modifications in the same oligonucleotide (e.g. phosphorothioate, 2'-O-methyl)?
Yes, we can synthesize oligonucleotides with mixed normal backbone / phosphorothioate groups or with mixed normal sugars / 2'-O-Methyl sugar and mixed phosphorothioate / 2'-O-Methyl sugars. Other combinations are also possible.
Is it possible to order a modification which is not in your catalog?
If the chemicals are commercially available, Eurogentec can modify your oligo with it. Please contact us
What is the longest RNA oligonucleotide you can synthesize?
RNA synthesis is not as straightforward as DNA synthesis. However, we have synthesized RNA 70 mers for customers and these have been used successfully.
What can I do to my PCR primer to change the mobility of the PCR product?
Addition of multiple thymidine residues at the 5'-end of a PCR primer will change the mobility of the PCR product. Large number of T residues can be added. An alternative is to use a non-coding hydrophilic monomer. The best choice is hexaethylene glycol (hexaethylene oxide). Multiple additions of this monomer can be carried out routinely.
Can unpurified oligonucleotides be used as PCR primers?
Yes, apparently so. Provided that the PCR primer is not very long (up to 30 bases) and the PCR product is not long, unpurified oligonucleotides will be efficient PCR primers. However, it is difficult to determine the quantity/concentration of a crude PCR primer because it will contain impurities such as protecting groups and short failure sequences that affect the UV spectrum. Hence, if the concentration of the primer is determined by UV absorbance (O.D. 260) you will not have as much of the full length primer in solution as you think. For this reason Eurogentec does not provide crude primers. Even Eurogentec "unpurified" oligonucleotides (Genomic oligos) are partly purified at no extra cost by SePop (Selective Precipitation Optimized Process) desalting. this ensures that short failure sequences and the largest part of contaminants have been removed.
What is the delivery time for oligonucleotide synthesis?
Standard desalted oligonucleotides (Genomic oligos) are normally synthesized on the day the order is received and sent out the next day. Modified oligonucleotides are synthesized and purified in a short timescale but the precise time depends upon the nature of the oligonucleotide.
For more information, check our delivery time table
Why using siRNA instead of other antisense oligonucleotides ?
The discovery of an efficient and easy way to knock out gene expression at the mRNA level has resembled the seek of the Holy Grail for more than 10 years. Unfortunately, the use of classical antisense techniques (i.e. using various chemically modified forms of oligonucleotides) has often resulted in insufficient or non-specific suppression of gene expression. It has recently been shown that the presence of short double-stranded RNA (siRNA for Small-Interfering RNA) induces RNA interference in most eukaryotic species, as well as in cell culture. RNA interference (RNAi) leads to the inhibition of protein expression via the sequence-specific, dsRNA-mediated destruction of target messenger RNA (mRNA).
It is believed that this discovery will have a tremendous impact on the study of molecular and cellular processes, and on gene function deciphering.
Why using synthetic siRNA instead of in vitro transcribed ones ?
In vitro transcription yields RNA molecules that are equally suitable for RNAi studies. Nevertheless, these experiments require expert skills at each step: in vitro transcription, purification, quantification... As a consequence, we frequently meet scientists irritated by the poor quality results they get after these tedious steps, and who have shifted to our user-friendly siRNA.
How does Eurogentec select target sequences ?
We use bioinformatics tools to select siRNA oligos with characteristics perfectly matching the last experimental results in this field.
Why using a 2 pyrimidines overhang at the 3' ends of my siRNA ?
Several studies have shown that the most efficient siRNA contains a dTdT (or UU) overhang at their 3' ends. Using dTdT is recommended for optimal stability of the siRNA duplex, however, UU overhangs work equally well.
How many siRNA should be purchased per target gene ?
It is advisable to test two to four siRNA sequences per gene. Nevertheless, in some cases, this number may vary from one to ten depending on your applications and particular needs.
What can I do if the designed siRNA doesn' t work ?
Despite its extreme efficiency, the selected siRNA might not work in your cell system. If so, it is advisable to check the following points:
- If no knock-out of the target gene is observed, it may be useful to analyze whether the corresponding mRNA was effectively degraded upon addition of the siRNA. Two or three days after transfection, the total RNA is extracted and subjected to further analysis. RT/PCR appears to be the method of choice since it is faster and far more sensitive than Northern blotting.
- Check for any sequencing error or polymorphism in your target gene. It has been shown that a single base mutation in the pairing region of the siRNA duplex is sufficient to abolish RNAi.
- Check that your cell line can effectively express the target mRNA.
What chemistry is used to synthesize Eurogentec's siRNA ?
Although similar to DNA synthesis, the additional 2'-OH group of RNA introduces considerable complexity to the RNA synthesis and requires a different protecting group.
All RNA oligos at Eurogentec (including siRNA) are made using monomers containing a 2'-O-tertiary-Butyl-dimethylsylil (TBDMS) protecting group, assuring robust and reliable synthesis.
All oligos are made with the standard phosphoramidite solid-phase synthesis technology.
In which form do I receive my siRNA ?
When you order one of our siRNA sets, you receive single-stranded siRNAs lyophilized in separate tubes, either HPLC-purified or desalted.
Each siRNA oligo set is supplied with RNase-free water for convenient resuspension.
Such conditioning may seem inconvenient, but it offers several advantages:
-
You can use each single oligo as a negative control.
-
You can test various combinations of modified strands.
Finally, even when ordered pre-annealed, it is highly recommended to perform a heat/cool step after resuspension to ensure proper duplex formation.
What kind of negative control can be used ?
Every researcher would tell it: "The choice of the right controls makes the whole difference between a good and a bad experiment". This adage is particularly true for RNAi studies.
Therefore, to maximize your result interpretation, the following precautions should be taken when using siRNAs:
- Always test the sense and antisense single strands in separate experiments.
- Try to use a scramble siRNA duplex. This should have the same nucleotide composition as your siRNA but lack significant sequence homology to any other gene (including yours). Some researchers use other types of negative controls such as : introducing 4 to 5 mismatches in the original sequence, flipping the middle four bases, or even reversing the entire sequence. In all cases, it is crucial to check that the negative control sequences will not silence another gene.
- If possible, knock-down your gene with two independent siRNA duplexes to control the specificity of the silencing process.
What cells can be used for RNAi studies?
Most readily transfectable cell lines are suitable for RNAi studies.
However, each new assay should be first optimized (i.e. by using a range of siRNA concentrations, by testing different (transfection reagent volume - siRNA amount) ratios ...) F
or a successful optimization, pay attention to the following points:
- Use only healthy cell cultures to assure good transfection reproducibility (i.e. don't use cells passaged more than 50 times).
- Try to avoid antibiotics during plating an up to 2 days after transfection.
RNA interference was first used in immortalized cell lines (such as the unavoidable Hela) whether adherent or in suspension. Recent studies indicate that even transfection resistant primary cell lines are suitable for RNAi research.
What is the typical extent of inhibition with Eurogentec's siRNA ?
We typically obtain 80 to 95 % inhibition at the mRNA level after 24 hours transfection. Inhibition at the protein level is highly dependent on the protein half life.
Why are my custom oligos delivered at ambient temperature?
Custom oligonucleotides shipped dried or in solution are stable for 2 weeks at room temperature when unopened and protected from light.
For long-term storage please check our stability statement.
How are my custom oligos shipped?
Custom oligonucleotides are shipped dried or in solution. They are stable for 1-2 weeks at room temperature when unopened.
For long-term storage please check our stability statement
Peptides
How should I store my peptides?
Lyophilized peptides
For the long-term storage of peptides, we recommend that peptides be stored as a solid powder. Lyophilized peptides can be stored at temperatures of -20 °C or lower with little degradation. Our catalog peptides are checked every 2 years from the date of QC release to ensure that these peptides are within the required purity specification. We recommend that customers also re-check their custom peptides every 2 years from the date of QC release.
Peptides in solution
Peptides in solution are much less stable and are susceptible to degradation. Solutions of water used to dissolve peptides should be sterile and purified.
In solution, some amount of the peptide may degrade depending on the amino acids present in the sequence. A few examples of which are shown below:
- Peptides containing methionine, cysteine, or tryptophan residues should have limited storage time in solution due to oxidation. These peptides should be dissolved in oxygen-free solvents.
- Glutamine and asparagine can deamidate to Glu and Asp, respectively
- Cysteines can undergo oxidative cyclization to form Cys-Cys disulfide bridges (intra or inter disulfide bridges can form).
- Charged residues (Asp, Glu, Lys, Arg, His) are hygroscopic (take up water from moisture in the air) and will easily form a viscous clear oil. This physical change may not affect the properties of the peptide. It is recommended that such peptides be first lyophilized to remove trace amounts of water, degassed with N2 to ensure that there is no water vapor in the vial, and quickly capped.
To prevent any damage caused by repeated freeze-thaw cycles, we recommend that the user only dissolves the amount of peptide needed for the immediate experiment. Any excess solutions should be stored at ≥ -20 oC until needed.
Avoid moisture
As moisture will greatly reduce the long-term stability of peptides, we recommend that the peptide should be allowed to equilibrate to room temperature in a desiccator before opening the vial. Once the peptide has been dispensed, any remaining peptide in the tube should be gently purged with anhydrous nitrogen, the container recapped, sealed with parafilm, and stored at -20 °C.
How do I solubilize my peptides?
Peptide solubility characteristics vary strongly from one peptide to another. Residues such as Ala, Cys, Ile, Leu, Met, Phe, and Val will increase the chance of the peptide being hydrophobic and dissolving in aqueous solutions. AnaSpec catalog peptides are tested for their solubility in certain solvents. This information is located on every QC Datasheet that is shipped along with the peptide. AnaSpec recommends that customers adhere to the below guideline for their custom peptides.
Solubility properties
Peptide solubility is highly dependent on the amino acid sequence. Hydrophobic peptides (high propensity of A, F, G, V, L, I, M, W, P) in nature, will require an organic solvent to dissolve. Acidic peptides (high propensity of D, E in the peptide sequence) require a basic aqueous buffer to dissolve, while basic peptides (high propensity of K, H, and R) require an acidic aqueous buffer to dissolve.
Selection of solvent
Taking into consideration the limitations of your assay, we recommend that the following guideline be used to determine the best solvent to dissolve your peptide
Hydrophobic peptides
To reconstitute a hydrophobic peptide, add 100 µL of DMSO and sonicate until a homogenous solution forms. Next, add your buffer of choice to form a 1 mg/mL solution (a higher concentration of peptide will require a greater amount of DMSO).
Hydrophilic (acidic) peptide
To reconstitute an acidic peptide, add 100 µL of 1% NH4OH to 1 mg of the peptide and vortex. After the formation of a clear solution, add your buffer of choice to form a 1 mg / mL solution.
Hydrophilic (basic) peptide
To reconstitute a basic peptide, add distilled water to the 1 mg of peptide and vortex.
What is the difference between net and gross peptides?
The industry standard is to deliver peptides in a lyophilized form and to state the delivery amount as the weight of the lyophilized powder “Gross weight”.
Gross weight contains the peptide of interest (including impurities), water, and a strong acid or base used in the preparative HPLC purification of the peptide. The majority of peptides are purified using trifluoroacetic acid (TFA). Water and TFA are byproducts of the purification process and there will always be trace amounts that cannot be removed completely.
TFA forms a salt with basic groups (Lysine, Arginine, Histidine, and free N-terminal amine) present in the peptide sequence. Usually, the minimum number of TFA molecules associated with a peptide is directly proportional to the number of basic residues in a given peptide sequence.
Therefore, the more basic residues present in a peptide, then the lower the actual amount of peptide present in the sample. To take into consideration the amount of TFA or water present in the sample, we recommend that the peptide content be determined for every custom peptide. The net peptide content (NPC) is the fraction of peptidic material present in the lyophilized material. In combination with the peptide purity, it allows one to determine the exact amount of the peptide of interest.
NPC is traditionally measured by amino acid analysis (AAA; limited accuracy but requires a low material amount) or elemental analysis (CHN; requires milligrams of peptide but is more accurate). Both methods measure total peptidic content.
Quant Peptides
We offer Quant-Peptides corresponding to two proprietary peptide quantitation methods (with and without Quant-Tag), offering net peptide content with better accuracy and reproducibility than AAA or CHN.
Could you help me for the design of my antigen peptide?
Thanks to our long experience in anti-peptide antibody programmes, Eurogentec has optimised the use of a combination of specific algorithms for peptide selection.
In our hands, the success rate of immunization using only one peptide per protein and after correct sequence analysis turns around 75 % statistically.
If the antibody production is carried out with two peptides out of the protein sequence, the failure risk will be reduced. The chance of success increases from 75% to about 90-95% (success = the protein is recognised in Western blot).
What type of chemistry do you use?
We utilize solid-phase Fmoc chemistry rather than BOC chemistry. Fmoc chemistry allows for the removal of protecting groups using mild conditions during peptide elongation, while BOC chemistry requires the use of a strong acid to remove the protecting groups during peptide elongation. Additionally, Fmoc chemistry utilizes trifluoroacetic acid (TFA) to remove the peptide from the resin, while BOC chemistry utilizes hydrofluoric acid (HF). HF is colorless, highly corrosive, and a contact poison.
Therefore, We employ Fmoc chemistry as it is more mild, flexible, and versatile compared with BOC chemistry.
What quality control (QC) information do you provide?
Our QC datasheet for catalog peptides includes:
- Testing attributes (appearance, % peak area by analytical HPLC, identity by MS, and solubility)
- Test method
- Acceptance criteria
- QC results
This information is listed on all of our QC datasheets and the raw data for each test can be provided upon request.
We can provide additional testing attributes as a fee for service offering.
How do I calculate molarity of my peptide?
Molarity refers to the molar concentration of a solution, which is the number of moles of solute dissolved in 1 liter of solution, expressed as mol/L, or M.
Molarity [M] = Mass / (Volume x Molar Mass); Mole = Concentration (g/L) x Volume (L)/MW (g/mol)
Example:
Given: 1mg of dry peptide powder with MW: 20KDa (Molar mass of peptide is 20 g/mol)
To determine molarity with known mass and known volume
For a 1ml solution of this peptide:
Molarity = Mass (0.001g) / (volume (0.001L) x Molar Mass (MW 20,000) = 50μM
To determine mass to achieve a certain molarity:
If for your assay, you need 0.01mM working peptide solution in 1ml of water, then calculate mass required as follows:
Mass = Molarity x Volume x Molar Mass
Mass = 0.01mM x (1/1000 L) x 20,000g/mol = 0.0002g
Hence, you will need 0.2mg/ml of peptide to have a working solution of 0.01mM.
How are the peptides supplied?
Peptides are shipped as lyophilized powder or oil at room temperature via an express courier. On request, we can also deliver your peptides in solution.
How do I know if my peptide will be soluble?
The solubility of a peptide in a given solvent is dependent on the amino acid sequence and often hard to predict. In the case of catalog peptides, the solubility information is listed on the QC datasheet. For custom peptides, please see the section entitled "How should I solubilize my peptide".
What spacer is there in a biotinylated peptide?
The standard biotin modification does not include a spacer. Spacers (PEG, X, etc.) are available on request. Please contact us for more guidance on the type of spacers that we offer.
How do you confirm a peptide is cyclized?
Generally, we can perform five types of cyclization reactions and these are shown below as:
- End to End [N-terminal to C-terminal]
- End to Side [N-terminal to a carboxylic acid from a side-chain residue]
- Side to End [Amine side-chain to C-terminal]
- Disulfide bridge cyclization
- Hydrocarbon-stapled peptides
End to End, End to Side, and Side to End cyclization products are usually confirmed by both a shift in molecular mass of 18 mass units in the Maldi-Tof mass spectrum as well as a change in the retention time in the analytical HPLC spectrum.
A disulfide bridge cyclization is confirmed usually by HPLC before and after the cyclization step. Although a mass shift of 2 mass units can be difficult to detect for certain peptides, a shift in the retention time of the analytical HPLC spectrum confirms that the peptide has been cyclized.
A hydrocarbon-stapled peptide consists of alkene that has been formed using a ruthenium catalyst. The hydrocarbon-staple ensures that the peptide conforms to an α-helix motif, which has shown to be useful for some therapeutic applications. This staple is confirmed by both a shift in the molecular mass (loss of 28 mass units) in the mass spectrum as well as a change in the retention time in the analytical HPLC spectrum.
Do you synthesize 'wobble' (or degenerate) peptide libraries?
We can synthesize ‘wobble’ or degenerate peptides. As there are known differences in the incorporation rates of different amino acids, we can compensate for these rates to produce roughly equimolar amounts of each ‘wobble’ peptide.
Do you synthesize isotope labeled peptides?
We can synthesize peptides containing non-radioactive atoms from commercially available building blocks. This includes amino acids containing 13C and/or 15N for Nuclear Magnetic Resonance (NMR) or Mass Spectrometry applications, just to name a few.
What is the difference between peptide content and peptide purity?
Peptide content is not an indication of the peptide purity as these are two separate measurements. Purity is determined by analytical HPLC and indicates the presence/absence of other peptidic contaminants (i.e. truncated peptides).
Net peptide content determines the actual amount of the peptide (peptide of interest as well as the peptide contaminants) present in the sample. The net peptide content is traditionally measured by Amino acid analysis (AAA; limited accuracy but requires a low amount of material) or elemental analysis (CHN; requires milligrams of peptide but is more accurate).
How do you generally purify your peptides?
Our peptides are generally purified by reverse-phase preparative HPLC, using 2 buffers containing 0.1% trifluoroacetic acid (TFA). The first buffer (Buffer A) is composed of 0.1 % TFA in deionized water and the second buffer (Buffer B) is composed of 0.1 % TFA in acetonitrile (ACN).
Generally, crude peptides are first dissolved in a solution of either Buffer A, some amount of Buffer B, or an organic polar solvent such as DMSO. If an organic polar solvent or Buffer B was used, this clear solution will first be diluted with Buffer A. Next, the peptidic solution is filtered and loaded onto the preparative HPLC system. Utilizing an optimized method gradient, fractions are then collected based on the absorbance readings. Each fraction is then analyzed via an analytical HPLC to ensure that the purity of each fraction meets the required purity specification.
These “pure” fractions are then pooled together and lyophilized. The “pure” powder is then tested for both purity and identity to ensure that the correct peptide has been manufactured.
Finally, an independent QC group at the company again tests the peptide to ensure that all of the testing attributes meet the required specifications.
What is Mass Spectrometry and why do we use it?
Mass spectrometry (MS) is an analytical tool used to determine the identity of molecules. In a typical MS procedure, a sample is ionized by bombarding the sample with a beam of electrons. The results are a plot of intensity as a function of the mass-to-charge ratio (m/z). These measurements can often be used to calculate the exact molecular weight of the sample components as well.
MALDI-TOF (Matrix-Assisted Laser Desorption Ionization - Time of Flight) Mass Spectrometry
MALDI-TOF is a technique that uses a pulsed laser that generates a laser energy absorbing matrix to create ions from large molecules with minimal fragmentation. MALDI-TOF instruments are often equipped with a reflectron that functions to increase the time of flight between ions of different m/z, which yields an increase in resolution.
ESI (Electrospray Ionization) Mass Spectrometry
ESI a technique used in mass spectrometry to produce ions using an electrospray in which a high voltage is applied to a liquid to create an aerosol. It is especially useful in producing ions from macromolecules because it overcomes the propensity of these molecules to fragment when ionized.
Can you explain the M+Na and M+K mass peaks in MALDI spectra?
It is very common to see Na (sodium) and K (potassium) adducts in the MALDI spectrum.
The sodium and potassium comes from the water used in the peptide solvents. Even distilled and deionized water has trace amounts of sodium and potassium ions, which can never be entirely removed. These become ionized during the MALDI mass spec process and bind to the free carboxyl groups of the peptide.
Because there is no water purification system that will remove every single sodium or potassium ion from water, seeing the sodium and potassium adducts at times is very common and unavoidable in MALDI mass spec. This is not an indication that the peptide is not pure, nor should it be confused with an incorrect molecular weight.
How long should a peptide that is going to be used as an antigen for antibody production be?
We usually use peptides that are 13 to 15 amino acids long in length, however shorter and longer peptides have been known to work.
We have successfully raised anti-peptide antibodies to peptides that differ in the presence/absence of a single phosphate (our phospho-specific antibody programme).
Antibodies typically recognise epitopes are typically between 6-8 amino acids, by presenting a peptide that is 15 amino acids long we increase the likelihood of generating useful antibodies but limit the chance of producing a peptide with a secondary structure that might be unrelated to the antigen.
What should I do with the ends of my peptides, keep them free or block them?
We recommend that the N-terminal and C-terminal ends of the peptide mimic the protein of interest. That being said, if the peptide sequence is derived from a sequence within the protein, then both the end of the peptide should be blocked. We recommend that the N-terminal end be blocked with an Acetyl group [Ac-NH2] while the C-terminal end is blocked with an Amide group [C(O)-NH2].
For peptide sequences that represent the N-terminus, We recommend keeping the N-terminus free (NH2) while the C-terminus of the peptide should be amidated [C(O)-NH2].
For peptide sequences that represent the C-terminus, We recommend keeping the C-terminus free (COOH) while the N-terminus of the peptide should be acetylated [Ac-NH2].
What methods do you recommend to conjugate a peptide to a carrier molecule?
The most important factor is to determine which end of the peptide should be conjugated to the carrier molecule. Once this has been determined, then there are a plethora of chemistries (linking agents) that can be used to conjugate the peptide to the carrier molecule.
Maleimide-Thiol Linkage
The preferred method for linking a peptide to the carrier protein is through the side chain thiol (-SH) of a cysteine residue located at either end of the peptide. Usually, the carrier protein is first modified using NBS (m-maleimidobenzoyl-N-hydroxysuccinimide ester). This reagent adds maleimide groups to the surface amines locate on the carrier protein. This maleimide-adduct is then reacted with the thiol group from the peptide and forms a new Sulfur-Carbon bond. The peptide is now conjugated to the surface of the protein via the maleimide linker.
Glutaraldehyde Linkage
Glutaraldehyde cross-links primary amino groups on the peptide to those on the carrier. This linkage is usually employed when there are no available Lys residues present in the peptide sequence. Thus, the N-terminal amine is used as the main point of conjugation.
Carbodiimide Linkage
Carbodiimides couple free carboxyl and amine groups, whether C- or N-terminal or on side chains (i.e lysine, aspartic acid, or glutamic acid) from the peptide to the carrier protein via an amide bond. Amide bonds are extremely rigid and this type of linkage results in considerable steric hindrance as the peptide is tightly bound and close to the carrier protein.
Peptides containing one or more lysine, aspartic acid, or glutamic acid can couple to the carrier protein at various places in the peptide giving alternative conjugates.
MAPS (Multiple Antigenic Peptides)
In some applications (whereby a protein carrier is not necessary) multiple peptide strands coupled to a Lysine backbone may be of great use. Typically 4, 8, or 16 copies of the peptide can be synthesized on a small multivalent core consisting of Lysine. The peptide is synthesized by the usual solid-phase peptide synthesis and cleaved from the resin to give the crude MAP-4, -8, or -16. As the molecular weight of the peptide is now increased several-fold, it may serve as a standalone and not require the linkage to a carrier.
How do I write my peptide sequence termini?
The international nomenclature used for the sequence termini is as follows:
- N-terminus: H means free amine (NH2-), Ac means acetyl [CH3C(O)-NH-], Pyr means pyroglutamic acid
- C-terminus: OH means free acid (-COOH), NH2 means amide [-CONH2]
- Modifications on the side chain of amino acids are depicted in the parenthesis after the corresponding amino acid. For example; phosphorylated serine = S(PO3H2) or epsilon-N-acetylated lysine = K(Ac)
What is a Pyroglutamyl peptide?
Pyroglutamyl (pGlu) peptides may spontaneously form when either Glutamine (Q) or Glutamic acid (E) is located at the sequence N-terminus. The conversion of Q or E to pGlu is a natural occurrence and in general it is believed that the hydrophobic γ-lactam ring of pGlu may play a role in peptide stability against gastrointestinal proteases. Pyroglutamyl peptides are therefore considered a normal subset of such peptides and are included as part of the peptide purity during HPLC analysis.
Protein, detection & labeling
What is the recomended buffer composition for protein electrophoresis using iD PAGE gels ?
iD Buffer composition
5x Sample Buffer
| SDS-PAGE | Native PAGE | Final concentration (1x Buffer) | |
| Tris base | 300 mg | 300 mg | 50 mM |
|---|---|---|---|
| Glycerol | 5.0 mL | 5.0 mL | 10% |
| Bromophenol blue | 25 mg | 25 mg | 0.01 % |
| 2-mercaptoethanol | 1.0 mL | 1.0 mL | 2% |
| SDS | 1.0 g | - | 2% |
| Deinonized water | to 10 mL | to 10 mL | - |
| pH (use 8M NaOH or 8M HCl) | 6.8 | 6.8 | - |
Store at room temperature
1x protein sample solution:
| Sample | x µL |
| Sample Buffer 5x | 2 µL |
| Deinonized water | to 10 µL |
Heat at 95°C for 10 minutes before loading on gel
10x MOPS Running Buffer: to separate medium to large proteins >20 kDa
| SDS-PAGE | Native PAGE | Final concentration (1x Buffer) | |
| Tris base | 60.6 g | 60.6 g | 50 mM |
|---|---|---|---|
| MOPS | 104.6 g | 104.6 g | 50 mM |
| EDTA | 3.0 g | 3.0 g | 1 mM |
| SDS | 10.0 g | - | 0.1 % |
| Deinonized water | to 1000 mL | to 1000 mL | - |
| pH (do not adjust) | 7.7 | 7.7 | - |
Store at 4°C up to 6 months
10x MES Running Buffer: to separate small proteins (2-50 kDa)
| SDS-PAGE | Native PAGE | Final concentration (1x Buffer) | |
| Tris base | 60.6 g | 60.6 g | 50 mM |
|---|---|---|---|
| MES | 97.6 g | 97.6 g | 50 mM |
| EDTA | 3.0 g | 3.0 g | 1 mM |
| SDS | 10.0 g | - | 0.1 % |
| Deinonized water | to 1000 mL | to 1000 mL | - |
| pH (adjust with 8M NaOH or 8M HCl) | 7.3 | 7.3 | - |
Store at 4°C up to 6 months
20x Transfer Buffer:
| 20x Transfer Buffer* | |
| Tris base | 60.6 g |
|---|---|
| Bicine | 81.6 g |
| Deionized water | 1000 mL |
To make 1L of 1x transfer buffer: mix 50 ml of 20x transfer buffer, 100mL of methanol or ethanol and 850 mL of deionized water.
qPCR/PCR Kits & Reagents
Is there an excess of reagent in the lyovial?
All dry products supplied in glass lyovials are designed to be dissolved in a volume and provide a 5% excess such that the customer can withdraw enough reagent to have a complete 1 mL final reaction volume. For example, if the mix is resuspended to provide a 2X master mix then the volume of water to be added is 525 uL.
What do I resuspend the cake with, buffer or water?
Dry MasterMixes are provided with a complete formulation; only nuclease-free water is required to resuspend the mixture as the product is already buffered.
Once dissolved how should I store the product I do not use?
Once dissolved the products may be stored short term in the fridge (2-8°C) for up to 24 hours, or in the freezer (-20°C) for up to 6 months. Up to 16%(v/v) glycerol (molecular grade) may be added to a 2X mixture to help reduce freeze thaw damage.
Do the Dry MasterMixes be temperatures sensitive during transport?
Dry MasterMixes can withstand transient excursions in temperatures up to 50°C during transport. Our recommended storage temperature for optimum product
performance and longevity is 15-30°C.
How do I store Takyon Dry MasterMixes?
Dry MasterMixes can be stored at ambient temperature. No cold supply chain is needed for transportion.
How can negative control be positive when detecting E.coli sequences?
If you are detecting bacterial sequences other than E.coli, the primers will have to exclude any part of the DNA that is shared between E.coli and the bacteria used (in other words, the primers have to be designed only in the part which is unique for the bacteria used).
If detecting E.coli sequences, There are some traces of bacterial DNA in the Taq polymerase and in the UNG (as it is produced in E.coli). You will have to determine the minimum level and subtract it from the positive signal. Everything above this signal should be considered as positive.
What can cause No Template Controls to give a positive result in the presence of UNG?
There can be an excess of the probe and the positive result is an artifact of this. Using a no amplification control where the Taq polymerase is left out can assess this. A positive result will then suggest an artifact and different reagent concentrations can be used.
What can cause No Template Controls to give a positive result?
The MasterMix may be contaminated with DNA template or PCR product from a previous PCR. Clean working practices should be used to avoid DNA template contamination. To avoid contamination from previous PCRs an UNG step should be introduced providing dUTP has been used in the dNTP mix.
All Eurogentec MasterMixes contain UNG, it can be ordered separately for the Core kits.
Does Eurogentec offer design for Real-Time probes?
Eurogentec is a specialist in the field of Real-Time PCR. Eurogentec offers a design service and can even do complete Real-Time projects.
Can Yakima Yellow™ be used with VIC settings?
As Yakima Yellow™ and VIC are very similar molecules. Yakima Yellow™ Double-Dye Oligos can be detected with VIC settings. As Yakima Yellow™ is normally combined with Eclipse® Dark Quencher, the background correction for TAMRA should be turned off (in plate set up window).
Can a FAM-TAMRA Double-Dye oligo be combined with a YakimaYellow™-Dark Quencher Double-Dye oligo?
It is possible to combine a FAM-TAMRA with a Yakima Yellow™- Eclipse® Dark Quencher Double-Dye Oligo. However, most Real-Time thermocycler correct for the background fluorescence of TAMRA, incase TAMRA is used as quencher. This means that the fluorescent signal of the FAM-TAMRA probe is measured correctly, but that the Yakima Yellow™-Eclipse® Dark Quencher signal is overcorrected and therefore gives to a lower sensitivity.
Therefore it is recommended to use dark quenchers when performing multiplex Real-Time PCRs, so lost of sensitivity can be reduced to a minimum.
Which fluorophores can be combined in a multiplex Double-Dye oligo assay?
It is dependent on the equipment which dyes can be combined. The combination that works best on most qPCR cyclers is FAM-Eclipse® Dark Quencher with Yakima Yellow™-Eclipse® Dark Quencher. Both are available from Eurogentec.
Which fluorescent dye should I use for my target and controls?
In most cases FAM is used for the target gene and Yakima Yellow™ for the control gene, when performing a multiplex Real-Time PCR.
Eurogentec does offer endogenous controls with both FAM and Yakima Yellow™.
How do typical cycling conditions for a Double-Dye oligo on a 96-well Real-Time cycler look like?
For most Double-Dye oligos, which have been designed using the Primer Express™ Software, used in combination with the qPCR MasterMix the following cycling conditions can be used:
50 °C 120s UNG hydrolysis step
95 °C 600s inactivation UNG, activation HotGoldStar
95 °C 15s denaturation } x 40
60 °C 60s annealing-elongation } x 40
Which probe systems can be used for allelic discrimination?
For allelic discrimination Double-Dye Oligos, Molecular Beacons, Scorpions® and hybridization probes and the most commonly used systems. The Molecular Beacons and Scorpions® have the advantage that they can be monitored at an optimal temperature, to make the distinguishing power as big as possible. The hybridization probes can be used together with melt curves and the Double-Dye Oligos are the easiest to use.
Which probe systems can be used for expression profiling?
All probe systems can be used for expression profiling. The Double-Dye Oligos are the most user friendly as they are easy to design, require minimal optimization and are easy to use.
Eurogentec offers Double-Dye Oligos that fit each qPCR cycler.
How can a low signal to noise ratio for Molecular Beacons be explained?
When designing Molecular Beacons, pay attention to the folding of the probe. It might fold into alternate conformations, which are not well quenched. Change the stem or loop sequence, or both to avoid this.
If the salt concentration of the buffer is too low (below 1 mM MgCl2) the probe does also not fold correctly.
Choose Eurogentec's Molecular Beacons and we will help you with the design.
How can a high background level be avoided when working with TAMRA, ROX, Cy® 3 or Cy® 5 as fluorophore in a Double-Dye oligo?
The commonly used quenchers TAMRA, DABCYL and Deep Dark Quencher I (DDQI) are not suitable to quench TAMRA, ROX, Cy® 3 or Cy® 5. For these long-wave emitting dyes Deep Dark Quencher II (DDQII) should be used. This quencher is available from Eurogentec.
What is the advantage of working with SYBR® Green I?
SYBR® Green I is an inexpensive, universal dye which binds to all dsDNA. It can be easily used in combination with a simple primer pair to detect PCR products in Real-Time. This dye is mainly very attractive for researchers analysis lots of different genes. However it is important to do a good primer design to avoid primer dimers, which will also be detected by SYBR® Green I.
What is the advantage of working with a probe system?
A probe system is always specific (except Amplifluor™ probes) and therefore does only detect the gene of interest. If you have a difficult to optimize PCR it will not show you any primer dimers or aspecific products.
With a probe system it is also possible to distinguish between similar sequences with small differences like SNPs or mutations.
In general, probe assays need less optimisation than SYBR® Green I assays.
Is a probe assay more sensitive than a SYBR® Green I assay?
A probe assay and a SYBR® Green I assay can be equally sensitive. In cases of difficult to optimize PCRs the SYBR® Green I assay might be less beneficial as it shows the total fluorescent signal of primer dimers, aspecific product and wanted product.
Can I use a SYBR® Green I kit for a probe assay?
This is not possible because the buffer for SYBR® Green I kits and the buffer for probe kits are different. The kits for SYBR® Green I applications contain special stabilizers to prevent the SYBR® Green I from degradation and do differ in pH and salt concentration.
What is the difference between a Core kit and a MasterMix?
A core kit is a 10x kit that contains all components in separate tubes, which gives a maximum in flexibility. The MasterMixes are 2x kits, with all components premixed. These guarantee a high reproducibility and ease of use. In average Core kits give a bit more sensitive results.
Eurogentec offers both types of kits.
Should the DNA and the qPCR MasterMix be mixed before thermocycling?
Spinning the 96-well plate or tubes will most of the time do, but the results are more reproducible if the plate or tubes are mixed on a magnetic shaker.
What is the difference between a one step and a two step RT qPCR reaction?
In a one step RT qPCR reaction the RT reaction and the qPCR reaction are done in one and the same tube. The buffer is a combination of a RT and a PCR buffer, in which both enzymes do work. The one step RT qPCR reaction is a closed tube assay, so contamination can be avoided. It saves pipeting steps and time and is easy in handling.
In a two step RT qPCR the RT reaction and the qPCR reaction are done in separate tubes. It gives a more flexible way of working in that the cDNA can be used for more than one qPCR reaction and can be archived, so that it eliminates the need to continually isolate RNA.
Further more it allows the use of oligo d(T) and ramdom nonamer primers in the RT step and specific primers in the PCR step. This will increase the specificity and sensitivity of the assay.
Eurogentec offers both one and two step RT qPCR kits.
In which case is one step RT qPCR recommended?
A one step RT qPCR is recommended when doing HTS (High Throughput Screening), where the experiment should be as easy and straight forward as possible and when doing experiments where all sources of contamination should be excluded.
Why is a Two step RT qPCR kit more efficient than a One step RT qPCR kit?
When performing a RT it is possible to use three different primer types:
Oligo d(T) primers, which bind to the poly A tail of the RNA and then only transcribe RNA. This will avoid contamination with genomic DNA. As the poly A tail is located at the beginning of the gene it will also lead to more full transcripts.
Random nonamers, which bind anywhere in the genome and allow the reverse transcriptase to fill up the gaps, will leads to high yields.
Specific primers, which bind to the gene of interest, and will therefore give specifics products.
The combination of oligo d(T) primers and random nonamers will give the highest yields and the longest transcripts, whereas specific primers transcribe only specific RNA but reduce the yield.
With an One step RT qPCR kit it is only possible to add specific primers, as it should be avoided that oligo d(T) primers and random nonamers participate in the PCR reaction, giving many aspecific products. As a RT reaction is performed at 40-50°C, the primers can bind with mismatches to the RNA and therefore transcribe unwanted sequences, which then also will be amplified in to consecutive PCR, leading to aspecific PCR products. This disadvantage is inherent to the method.
In a Two step kit the oligo d(T) primers and the random nonamers are included in the kit and will give ride to cDNA. Then two specific primers have to be selected and this will amplify the exact sequence.
Why do qPCR kits contain dUTP (and UNG)?
qPCR kits contain dUTP and UNG to prevent carry over from previous qPCRs.
The kits contain normal dNTPs, mixed with dUTPs. The dUTPs will be incorporated instead of dTTPs during the qPCR. UNG (Uracil-N-glycosylase) is an enzyme, which hydrolyses the release of free uracil from uracil containing DNA. When the qPCR reaction is started with an initial incubation with UNG, no contamination through carry over from previous qPCRs will be seen, as that DNA will be degraded, so it cannot participate anymore in the PCR process.
Why does an initial step at 50 °C during 2 min and a second step at 95 °C during 10 min have to be performed when using UNG for carry over prevention?
When performing an assay with UNG, a first step at 50°C during 2min and a second step at 95°C during 10min has to be added before performing a normal qPCR:
50 °C during 2min
95 °C during 10min
94 °C during 15s
55 ºC-65 ºC during 30s
72 ºC during 60s
repeat during 35 cycles
Hold at 50 °C for ever
The fist step is needed to activate the UNG enzyme, to allow it to degrade U containing ds DNA.
The second step at 95 °C will deactivate the UNG and will activate the HotGoldStar.
Why do certain qPCR kits contain ROX passive reference?
All kits meant for the use on the ABI Prism™ 5700, 7000, 7700 and 7900 contain a ROX passive reference as these systems require a constant ROX level to be able to calculate the fluorescent signal (ΔRN) correctly.
How do I avoid amplification of genomic DNA, which can be a contamination factor in my isolated RNA in RT qPCR?
By designing exon-exon spanning primers you will only amplify cDNA, as the primers do not bind to gDNA. In this way your amount of PCR products is directly related to the initial amount of cDNA, which is again directly related to the initial amount of RNA.
What can cause a SYBR® Green I reaction, which has been working well, stop working?
SYBR® Green I is unstable if diluted in a watery solution and also if it is not kept in the dark. Therefore Eurogentec offers kits that contain DMSO to stabilize the SYBR® Green I. Kits should be stored in the dark.
How to optimize a qPCR assay?
If the primers and Double-Dye Oligo have been designed using Primer Express™ and are used in combination with the qPCR MasterMix or qPCR MasterMix for SYBR® Green I the following matrices can be used to define the correct primer concentration and the correct probe concentration. First define the optimum for the primers and then determine the best concentration for the probe that fits to the primer optimum.
Are DNA or RNA standards preferred in qPCR?
DNA standards are preferred when starting from genomic DNA. RNA standards are preferred when starting from RNA because then the efficiency of the RT reaction can also be measured.
What is UNG and how does it work?
UNG stands for Uracil-N-glycosylase. It is an enzyme, which hydrolyses uracil-glycosidic bonds in DNA containing dUTP, therefore degrading the DNA into small fragments. In this way contamination from previous qPCR reactions can be avoided.
Is there a difference in PCR efficiency when using dUTP instead of dTTP?
dUTP is not naturally present in DNA and therefore not preferred by the Taq polymerase. The Taq polymerase will incorporate dUTP, but not with the same efficience as normal nucleotides. Therefore the efficiency of the reaction is reduced compared to a reaction in which on dATP, dCTP, dGTP and dTTP are used.
Eurogentec offers kits that contain no dUTPs. However, these kits have to be used very carefully, as there will be no possibility to digest the PCR products with UNG.
How to select a good endogenous control?
A good endogenous control is not up regulated by the effect that needs studying, so the condition, drug, therapy, medium etcetera. It should be expressed in the same range and there should preferably be no similar genes or pseudogenes of this gene.
Cytoskeletal molecules are often a good choice.
What is the difference between endpoint analysis and Real-Time analysis?
qPCR can be done both as endpoint analysis and Real-Time analysis. In endpoint analysis the fluorescence (ΔRn) is measured when the PCR is complete. In Real-Time analysis the fluorescence is measured in each cycle of the PCR. It is only possible to quantify in real time, as the final yield of PCR products is not proportional to the initial amount of DNA, due to limiting factors at the end of the PCR. Therefore endpoint analysis is often used for allelic discrimination and, Real-Time analysis is used for quantification.
How can the performance of allelic discrimination be increased?
The probe design might be bad or the mismatch created is particularly stable. Try redesigning the primers/probe. It may be worth trying to place the primer over the mutation rather than the probe if a design is particularly difficult. A melt curve can help to indicate at which temperature the best discrimination is obtained. An additional detection step can be programmed in the PCR protocol.
Eurogentec offers a design service for qPCR probes.
What is absolute quantitation?
Absolute quantitation is a method in which unknown samples are quantitated by comparing them to a standard curve, based on absolute amounts of DNA.
What is relative quantitation?
Relative quantitation is a method in which unknown samples are compared to reference samples to determine the increase or decrease of gene expression. The out come is presented as an index, not an absolute amount.
In which buffer should qPCR probes be resuspended?
As fluorophores are sensitive to hydrolysis, Eurogentec recommends resuspending qPCR probes in TE 0.01 instead of just water. If the qPCR probes are desolved in an acidic solution the fluorophores can hydrolyze and will give a lower signal to noise ratio.
How should the qPCR and RT qPCR kits be stored?
For long term storage the Takyon™ qPCR MasterMixes and Core Kits should be stored at -15 °C to -25 °C in a constant temperature freezer. When stored under these conditions the reagents are stable during 12 months (MasterMixes) or 24 months (Core Kits).
For short term storage the Takyon™ qPCR MasterMixes and Core Kits can be stored at 4 °C for 6 months.
The Takyon™ Dry MasterMix dTTP can be stored at ambient temperature (15-35°C) up to 18-months. This is valid for unopened bottles (inert gas inside for long term stability)
!!! Do not expose the dried reagent to light as ROX normalisation dye is light sensitive
What can cause a very high background level when working with probes?
Probes can be degraded separating the fluorophore from quencher, leading to a high background level of the fluorophore. When aliquoting the oligonucleotides, sterile tubes and tips must be used to avoid contamination with DNases.
How can a low signal to noise ratio for probes be explained?
The most likely reason is contamination by either free fluorophores or oligonucleotides that contain the fluorophore but not the quencher. The fluorophores can be removed by HPLC.
All Eurogentec probes for Real-Time PCR are HPLC purified, Mass Spectrometry controlled and HPLC controlled.